
運動神経障害
最後に見直したもの: 12.07.2025

すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
神経障害の中でも運動神経障害または運動ニューロパチーは、神経系のさまざまな構造によって提供される反射運動機能の障害として定義されます。
運動の病理には、神経インパルスの伝導に関与する皮質下運動核、小脳、錐体系、脳幹網様体、骨格筋を支配する末梢神経、運動ニューロンとその突起(軸索)の損傷が関与している場合があります。
疫学
臨床統計によると、糖尿病患者の 10 人中 6 人は時間の経過とともに末梢運動神経障害を発症します。
Journal of Neurologyによると、ヒト免疫不全ウイルスは患者の3分の1に神経障害を引き起こします。多発性運動神経障害は人口10万人あたり3人に認められ、男性では男性のほぼ3倍の頻度で発症します。
末梢神経の最も一般的な遺伝性疾患であるシャルコー・マリー・トゥース病は、約 2500 ~ 5000 人に 1 人が罹患します。
北米では、脊髄性筋萎縮症は毎年6,000人から8,000人に1人の乳児に発症します。ある推定によると、40人から50人に1人がこの疾患の無症候性キャリアであり、これは常染色体優性遺伝として子供に受け継がれる可能性のある欠陥遺伝子を持っていることを意味します。
原因 運動ニューロパチー
運動神経障害の原因は多くの場合、運動ニューロン疾患です。これらの神経細胞は、脳内の上部と脊髄内の下部に分かれており、前者は脳の感覚運動皮質の核から脊髄へ神経インパルスを伝達し、後者はそれを筋線維のシナプスへ中継します。
上位運動ニューロンの変性変化の場合、原発性側索硬化症および遺伝性痙性対麻痺が観察されます。脊髄運動ニューロンの変性病変の場合、局所性脊髄運動ニューロン症候群または筋萎縮性側索硬化症候群、多発性運動神経障害および遠位脊髄筋萎縮症が発生します。病因的に均一な症候群も区別されます。Werdnig-Hoffman症候群(生後6か月までの乳児に発症)、Dubowitz症候群(生後6~12か月で発症)、Kugelberg-Welander症候群(2~17歳に発症する)、フリードライヒ運動失調症(10歳代後半までに発症)などです。成人では、最も一般的なタイプの脊髄筋萎縮症は、緩徐進行性ケネディ病(脊髄延髄性筋萎縮症とも呼ばれる)と考えられています。
運動神経障害は、筋緊張と運動協調を制御する小脳の変性と関連しています。遺伝性運動障害または運動失調症、多発性硬化症の症状、急性脳血管障害の神経学的結果として発症します。詳細については、「虚血性神経障害」をご覧ください。
運動障害は腫瘍性疾患、特に腫瘍随伴性神経症候群(イートン・ランバート症候群)において発生することがあります。「小脳性運動失調症の原因」を参照してください。
外傷性脳損傷やさまざまな毒性物質による中毒を患った患者、ポリオーマウイルス、水痘ウイルス、帯状疱疹ウイルス、ヒト免疫不全ウイルス(HIV)、サイトメガロウイルス、およびボレリア・ブルグドルフェリ菌、マイコプラズマ・ホミニス菌、カンピロバクター・ジェジュニ菌、トレポネーマ・パリダム菌(神経梅毒の原因菌)などの感染症にかかった患者は、神経学的合併症として運動機能障害を呈することがよくあります。
危険因子
運動神経障害の発症の危険因子として、専門家は自己免疫反応の活性化を伴う免疫系の障害、神経線維のミエリン鞘および運動ニューロンの軸索の喪失を挙げています。
リスクが高いのは、高齢者、アルコール依存症者、遺伝性神経疾患を持つ家系の子供、リンパ腫や肺がんの患者、電離放射線や細胞増殖抑制剤を用いたがん治療後の患者です。詳細は「化学療法後の多発神経障害」の記事をご覧ください。
糖尿病患者における神経運動障害のリスクは非常に高い。糖尿病における運動神経障害については、「糖尿病性神経障害」という出版物で詳細に論じられている。
運動神経合併症を伴うその他の疾患としては、セリアック病、アミロイドーシス、巨赤芽球性貧血(ビタミン B12 欠乏症)、および狼瘡(SLE)などがあります。
感覚運動障害を引き起こす可能性のある薬剤を使用する際には、そのリスクを考慮する必要があります。例えば、ジスルフィラム(アルコール依存症治療薬)、フェニトイン(抗てんかん薬)、抗がん剤(シスプラチン、ビンクリスチンなど)、高血圧治療薬アミオダロンなどがこれに該当します。
病因
遺伝性脊髄性筋萎縮症の場合、病因は脊髄運動ニューロンおよび脳幹の一部の変性にあります。これは、運動ニューロン核のSMNタンパク質複合体をコードするSMN1遺伝子(5ql3座位)の変異によって起こり、そのレベル低下が運動ニューロン細胞の死につながります。筋萎縮は、脳神経の運動ニューロン核および効果神経終末(神経筋シナプス)の欠陥によって発症する可能性があります。筋力低下(筋緊張の低下、腱反射の弱化、場合によっては萎縮)は、運動ニューロンの軸索からシナプス間隙へのアセチルコリンメディエーターの放出が制限される結果である可能性があります。
免疫学的に条件付けられた神経障害の病態生理学的メカニズムには、異常な細胞性免疫応答および体液性免疫応答が含まれます。したがって、多巣性および軸索性運動神経障害の発症は、運動ニューロンおよび末梢神経の軸索のミエリン鞘に対するIgM抗体の形成に関連しています。ミエリンには、スフィンゴ脂質とオリゴ糖の複合化合物であるガングリオシドGM1、GD1a、GD1bが含まれています。ガングリオシドGM1に対して特異的に産生された抗体がGM1と相互作用し、補体免疫系を活性化し、イオンチャネルを遮断すると考えられています。運動ニューロンの軸索鞘におけるGM1のレベルは感覚ニューロンの鞘よりも高いため、運動神経線維は自己抗体による攻撃を受けやすいと考えられます。
また、慢性炎症性脱髄性神経障害についても読んでください。
症状 運動ニューロパチー
運動神経障害の種類によって、運動機能障害の特徴である特定の類似した症状が現れます。
初期症状は、病気の部位と病因によって決まります。例えば、筋萎縮性側索硬化症の初期症状は、腕と脚の進行性の筋力低下と硬直として現れ、歩行障害(運動協調とバランスの障害を伴う歩行の遅延、平地でもつまずくことが多い)を引き起こします。
成人の遺伝性脊髄性筋萎縮症では、運動神経障害の症状として、表層筋線維の周期的なけいれん(線維束性収縮)が、筋緊張の低下と腱反射の弱化を背景に現れます。病気が進行する後期には、腕や脚だけでなく、他の筋肉群(肋間呼吸筋、咽頭筋、口腔顔面筋など)にも、動きを制限する筋力低下が感じられ始めます。このため、呼吸困難が生じ、呼吸不全や嚥下障害(飲み込みにくい)へと進行します。発話も遅くなり、不明瞭になります。脊髄と脳幹の運動ニューロンの遺伝的に決定された変性を伴うケネディ病の典型的な症状には、四肢、顔面、咽頭、喉頭、口腔の筋肉の筋力低下と萎縮が含まれます。発話障害(構音障害)と嚥下障害(嚥下障害)が認められます。
多発性または多巣性運動神経障害は、感覚症状を伴わない片側性四肢運動障害として発症します。10例中8例が40~50歳で発症します。尺骨神経、正中神経、橈骨神経が最も多く侵され、手と手首の筋力低下により微細運動能力が阻害されます。ルイス・サムナー症候群は、本質的に後天性(炎症性)の多発性運動感覚神経障害であり、上肢の知覚異常と手の甲の皮膚感覚低下を呈します。詳細は「上肢の神経障害」の記事をご覧ください。
下肢の運動神経障害は、広く普及している神経疾患の一種であり、出版物「脚の神経障害」で詳細に議論されています。
免疫学的に誘発される神経障害には、急性型と慢性型があります。神経突起の機能不全によって引き起こされる急性軸索運動神経障害は、ランドリー・ギラン・バレー多発神経炎またはギラン・バレー症候群(急性多発神経根炎)の亜型または変異型として診断されます。症状としては、上肢末端部の進行性筋力低下、線維束性収縮、部分的な腱反射消失、眼球運動制限、神経インパルス伝導ブロックを伴わない弛緩性四肢麻痺(四肢麻痺)などが挙げられます。この病態では、脱髄や感覚障害の兆候は認められません。
慢性特発性軸索運動性多発神経障害は、高齢者(65歳以上)によく見られる神経疾患で、足首クローヌス、歩行時の筋力低下および硬直、安静時のふくらはぎの筋肉の痛みを伴うこむら返り(こむら返り)、歩行後の前脛骨筋のこむら返りといった、下肢の左右対称の末梢症状を呈します。
運動ニューロンの突起のミエリン鞘(および筋肉を支配する脊髄神経の根と線維)の個々の部分の破壊につながる病理学的プロセスにより、運動軸索脱髄性ニューロパチーが発症することがあります。症状としては、四肢の筋肉の不随意なけいれん、感覚異常(チクチク感や痺れ)、触覚および温度感覚の低下(特に手足)、麻痺(部分的な麻痺)、下半身麻痺(両腕または両脚の同時麻痺)、起立性めまい、歩行障害、構音障害などが挙げられます。栄養症状としては、発汗の増加や心拍数の上昇/低下などが挙げられます。
フォーム
遠心性(運動)および求心性(感覚)ニューロンと神経線維が信号を伝達する能力を失うと、遺伝性神経障害を持つ小児および青年に最も多く発生し、末梢運動感覚神経障害と診断されます。これは遺伝的に決定される疾患のいくつかの種類に分けられます。
運動感覚ニューロパチー 1 型(肥厚性脱髄性)は、小児の遺伝性末梢ニューロパチーの半数を占め、染色体 17p11.2、1q21-q23、および 10q21 の遺伝子変異によるミエリンタンパク質の合成障害によって引き起こされる分節性脱髄に関連しています。
末梢神経の肥大を特徴とするこのタイプの病理は、下肢の腓骨筋(腓骨筋)のゆっくり進行する萎縮です - シャルコー・マリー・トゥース病 1 型。足首領域の膝下の脚の筋肉の萎縮を特徴とし (病的に高い足のアーチの形成と足指の形状の特徴的な変化を伴う)、緊張に伴って震えが起こることが多く、無汗症 (発汗の欠如) と進行性の知覚鈍麻が認められ、場合によっては痛覚の喪失 (下肢の遠位部)、アキレス靭帯の腱反射の消失、精神および精神障害の兆候の出現、まれに神経性難聴を伴うことがあります。後期には、肘下の腕の筋肉も萎縮し、手の変形を伴います。
遺伝性運動感覚ニューロパチー2型(シャルコー・マリー・トゥース病2型)は、軸索性筋萎縮症であり、ミエリン鞘の喪失を伴わずに運動ニューロンと感覚ニューロンの突起の機能不全および変性を伴う疾患です。同じ筋群が侵され、5歳から25歳の間に発症します。染色体1p35-p36、3q13-q22、および7p14に変異が同定されています。
神経インパルス伝導速度はほぼ正常(疾患の第1型と比較して)であるため、遠位筋の筋力低下および萎縮の臨床症状は比較的軽度です。膝下筋の萎縮は患者の75%で対称性を示します。典型的な初期症状は、足と足首の筋力低下、腱反射の低下、足首背屈筋の筋力低下です。軽度の感覚症状が認められ、疼痛、睡眠時無呼吸、むずむず脚症候群、抑うつ症状が認められる場合があります。腕の筋萎縮はまれです。
合併症とその結果
かつて神経科医は、運動ニューロン疾患は脳の機能に影響を与えないと考えていましたが、研究結果により、この見解は誤りであることが示されました。筋萎縮性側索硬化症および下位運動ニューロンの変性変化による悪影響や合併症は、患者のほぼ半数に何らかの中枢神経系障害として現れ、15%の症例では前頭側頭型認知症を発症することが判明しました。また、制御不能な泣き声や笑い声を伴う性格や感情の変化が現れることもあります。
筋萎縮性側索硬化症では、主な呼吸筋(横隔膜)の収縮障害により呼吸障害が起こり、患者は不安や睡眠障害も増加します。
軸索脱髄型神経障害の合併症は、腸管運動障害、排尿障害、勃起不全として現れます。
感覚神経が損傷すると、痛みに対する感受性が失われる可能性があり、感染性炎症による負傷や傷を治療せずに放置すると、壊疽や敗血症につながる可能性があります。
シャルコー・マリー・トゥース病では、関節が圧力に正常に反応できず、骨構造に微小な亀裂が生じ、骨組織の破壊により手足が不可逆的に変形します。
脊髄性筋萎縮症は、世界で2番目に多い小児死亡原因と考えられています。病変の程度が軽微であれば、患者は生存しますが、多くの場合、その後、自立した運動能力を失います。
診断 運動ニューロパチー
神経運動障害は、初期段階では、症状が多発性硬化症、神経炎、パーキンソン病などの他の病気の症状と似ているため、診断が困難です。
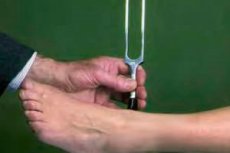
診断は腱反射の検査と検査から始まります。生化学検査、一般血液検査、血漿中のクレアチニンホスホキナーゼ値、C反応性タンパク質、抗体値(特にガングリオシドGM1に対する抗体)、補体C3などの検査が必要です。必要に応じて、脳脊髄液検査も行います。
神経学で使用される主な機器診断には、刺激筋電図検査 (EMG)、電気筋電図検査 (ENMG)、脊髄造影検査、脳の超音波および MRI スキャン (脳卒中、脳腫瘍、循環障害、構造異常を除外するため)、陽電子放出断層撮影 (PET) などがあります。
一部の運動神経障害は筋萎縮性側索硬化症の亜型に分類されますが、鑑別診断が必要です。髄鞘破壊を伴う免疫起源の神経障害の中では、多巣性運動神経障害と慢性免疫脱髄性多発神経障害を鑑別する必要があります。
感覚神経に関わる下位運動ニューロンの喪失は、腫瘍随伴性脳脊髄炎や感覚神経節症候群と区別する必要がある。
さらに、脊髄を視覚化する脊椎のMRIを使用して、筋肉の研究が行われるミオパチー症候群と筋ジストロフィー、およびモルバン病(脊髄空洞症)を除外する必要があります。
連絡先
処理 運動ニューロパチー
神経科医は、運動神経障害の治療は現状では対症療法のみであり、患者の症状を緩和し、病態の進行をある程度遅らせることは可能だと認めています。また、遺伝性の運動神経障害および感覚神経障害を治療する薬剤はまだ存在しません。
一般的に受け入れられている方法の 1 つは、患者の血液から自己抗体を除去する定期的な血漿交換です。
多発性運動神経障害では、ヒト免疫グロブリン(IVIg)を点滴で投与します。また、免疫調節作用を持つグルココルチコイド(プレドニゾロンまたはメチルプレドニゾロン)を全身投与することも可能です。ビタミンA、D、およびビタミンB群は、あらゆる種類の運動障害に処方されます。
他の薬剤も使用されます。まず、L-カルニチンは、組織の代謝プロセスを正常化し、損傷した細胞を修復するために経口投与されます。成人の場合はカプセル(1日2回、0.25~0.5g)、小児の場合はシロップ(投与量は年齢に応じて医師が決定)です。
神経インパルスの伝導性を高めるために、コリンエステラーゼ酵素の阻害薬であるイピダクリン(他の商品名:ニューロジミン、アミピリン、アキサモン)を経口または非経口で使用します。成人は1回10~20mgを1日3回(または筋肉内投与で1ml)服用します。1歳から14歳までの小児は経口投与のみが許可されます。1回投与量は10mg(半錠)で、1日3回まで服用できます。治療期間は1~1.5ヶ月です。イピダクリンの再投与は、初回投与終了から2ヶ月後に可能です。
この薬は、心拍障害、炎症性消化器疾患および肺疾患、および妊娠には禁忌です。また、最も起こりやすい副作用としては、吐き気、下痢、めまい、流涎、気管支痙攣などが挙げられます。
最近、海外の神経科医が、筋萎縮性側索硬化症(ALS)の患者に、新薬(FDA承認)リルゾール(リルテック)を処方しています。その有効性や作用機序は未だ十分に解明されておらず、その使用に伴う合併症のリストには、深刻な副作用が数多く含まれています。
資料からの有用な情報 -糖尿病性神経障害の治療と出版物 -上肢神経障害の治療
遺伝性運動感覚障害の治療には、神経科医だけでなく理学療法士の関与も必要です。理学療法は、病気の進行を遅らせ、予防し、症状を管理する上で重要な役割を果たします。治療計画では、障害のある筋群の強化に重点を置く必要があります。具体的には、治療マッサージ、運動療法、超音波療法、電気刺激療法、水療法、ペロセラピーなどが挙げられます。
多くの患者は整形外科的支援を必要とします。歩行時にアーチを支えるために整形靴や足首・足の矯正具が必要です。また、松葉杖、杖、歩行器が必要になる場合が多く、車椅子が必要な患者もいます。
重度の四肢変形の場合は外科的治療が行われます。
民間療法を好む人には、蜂毒の使用が推奨されます。蜂の刺し傷による治療です。
しかし、ミツバチ毒(その有効成分メリチンを含む)の有効性は、化学療法によって引き起こされる末梢神経障害に対してのみ証明されていることを心に留めておく必要があります。
しかし、カモミールとラベンダーのエッセンシャルオイル(メインオイルをデザートスプーン1杯につき数滴)を使ったマッサージは、運動障害の知覚異常に効果があります。
同様に、抗がん剤の使用によって引き起こされる神経障害にもハーブ療法は有効です。以下の薬用植物が用いられます。
- セイヨウサルビア(Salvia officinalis)には、強い抗酸化作用があり、末梢神経系の神経細胞を保護するアピゲニンが含まれています。
- ショウブ(Acorus calamus)、その抽出物は痛みを和らげ、鎮静させ、けいれんを和らげる。
- イチョウ(Ginkgo biloba)には、損傷したニューロンに良い効果をもたらすテルペントリラクトンが含まれています。
進行性脊髄筋萎縮症の場合、ホメオパシー療法も用いることができ、以下の製剤が推奨されます:アルゲントゥム・ニトリカム、プルンバム、フォスフォラス、カリ・フォスフォリカム、クプラム、アルニカ・モンタナ。しかし、これらの製剤は、運動機能障害を引き起こす遺伝的に「プログラムされた」病態には効果がありません。
予測
病気の発症の予後は、運動神経障害の原因、神経信号の伝導を保証する構造への損傷の程度、および中枢神経系の反射運動機能によって異なります。
多くの場合、これらの病気は急速に進行し、機能的欠陥が非常に重大であるため、患者は障害を負うことになります。
遺伝性の運動感覚神経障害は平均寿命を縮めることはありませんが、関連する病状により神経障害のさまざまな合併症が発生します。
 [ 39 ]
[ 39 ]