
脳の多毛症
最後に見直したもの: 04.07.2025

すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
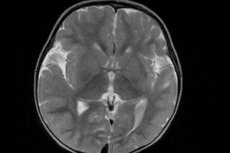
先天性欠損症(大脳皮質の細胞構造の全体的な変化を伴い、異常に小さな脳回が多数形成される)は、脳多小回(ラテン語のgyrus(回旋)に由来)と定義されます。[ 1 ]
疫学
統計によると、あらゆる種類の脳形成不全のうち、皮質の先天異常は症例の約3分の1に認められますが、単独の多小脳回の発生率に関するデータはありません。
原因 多精子症
多小脳回症の具体的な原因はまだ調査中ですが、その病因の本質は、脳の発達における他のすべての欠陥と同様に、胚の発達における逸脱にあります。[ 2 ]
この場合、胎児脳の回転過程、すなわち大脳皮質に特徴的なひだの形成が阻害されます。このひだの形成は妊娠中期頃から始まり、ひだの頂点から脳回が形成され、ひだの間の窪みから脳溝が形成されます。頭蓋内のスペースが限られている状況では、脳回と脳溝の形成によって大脳皮質の面積が増加します。[ 3 ]
大脳皮質の子宮内発育障害は、ほとんどの場合、染色体異常や遺伝子変異によって引き起こされます。これは、1つの遺伝子の変異の場合もあれば、隣接する複数の遺伝子の欠失の場合もあります。[ 4 ]
多小脳回は単独で発生することもありますが、他の脳異常、特に遺伝的に決定される症候群、特にディジョージ症候群(22q11.2染色体欠失症候群)を伴って発生することもあります。[ 5 ] アダムズ・オリバー、ゼルウィガー、ウォーカー・ウォーバーグ症候群、アイカルディ症候群(脳梁無形成症)、スミス・キングスモア症候群(大頭症)、ゴールドバーグ・シュプリンツェン症候群(小頭症および顔面異形症)などです。[ 6 ]、[ 7 ]
危険因子
専門家は、多小脳回症の発症の危険因子として以下の点を考慮しています。
- 遺伝性の遺伝子欠陥;
- 胚における自然発生的な遺伝子変異。
- 妊娠中のサイトメガロウイルス感染を中心とした毒素や感染症による胎児への悪影響。
- 胎盤灌流不足と胎児の酸素欠乏による脳虚血;
- 様々な原因による胎児硬膜下出血[ 8 ]
病因
脳回形成の生理学的メカニズムは今日まで解明されていない(複数の説がある)にもかかわらず、多小脳回症の病因は、神経堤の胎児細胞(神経芽細胞)の移動、分裂、増殖を含む脳構造の神経発生の障害と関連している。また、既に述べた胎児脳の脳回形成の障害とも関連している。
これらの疾患の結果、脳の結合組織膜(軟膜)とクモ膜(クモ膜)に欠陥が生じ、層の厚さや数の変化、隣接する脳回における分子層の融合、膜の血管新生の増加による脳灌流障害(軟部皮質の局所出血、その下の白質の浮腫、皮質の一部の萎縮も起こり得る)がみられる。[ 9 ]
大脳皮質の組織形成において、その軟膜である基底膜は重要な役割を果たします。研究により、多小脳回やその他の皮質異常は、この膜の不安定な成長と、そのタンパク質および糖タンパク質成分(IV型コラーゲン、フィブロネクチン、ラミニンなど)の欠陥に関連していることが示されており、これが皮質の細胞構造の病理学的変化につながります。
多小脳回において変異が同定されている遺伝子の中には、例えば染色体16q21上のGPR56(またはADGRG1)遺伝子が挙げられます。この遺伝子は、細胞接着受容体の膜Gタンパク質をコードしています。細胞接着受容体は、胚の形態形成過程を制御し、形成される組織の形態を決定する細胞間接着部位です。この遺伝子の変異は、両側性前頭頭頂葉型多小脳回の発生と関連しています。[ 10 ]
症状 多精子症
小児の脳の片側が多小脳回を呈する場合は片側性、両半球の皮質が影響を受ける場合は両側性と呼ばれます。多小脳回という皮質奇形は、主に背外側皮質を侵します。
最初の兆候と時間の経過とともに現れる臨床像は、異常によって脳のどの特定の領域が影響を受けるかによって完全に異なります。
片側性局所性多小脳回は、脳の比較的狭い領域を侵し、前頭葉または前頭頭頂葉皮質、およびシルビウス溝(外側溝)付近のシルビウス溝周囲皮質に広がることが最も多い。発作として発現し、他の神経症状は見られない場合もあります。
両側性多小脳回症の症状には、再発性てんかん発作、発達遅延、筋力低下、斜視、嚥下障害(嚥下障害)および発話障害(構音障害)などがあります。
したがって、頻繁な発作に加えて、両側前頭多小脳回は、子供の全般的および精神的発達の遅れ、痙性四肢麻痺(下肢と上肢の弛緩性麻痺)、運動失調(運動協調の障害)、歩行障害(歩行障害)、そして多くの場合、運動失調(完全に立つことができない)および歩行不能(歩くことができない)として現れます。
前頭頭頂葉多小脳回または両側前頭頭頂葉多小脳回は、発達遅滞、認知障害(中等度または重度)、発作、視線共同の欠如および斜視、運動失調、筋緊張亢進などの症状を特徴とする。[ 11 ]
両側性シルビウス膜周囲多小脳回がある場合、症状(出生時、乳児期、または2~3歳頃に現れる)の中で最も頻繁に観察されるのは、四肢のけいれんや痙縮、嚥下障害や流涎、顔面、舌、顎、喉頭の筋肉の部分的な両側麻痺、および全般的および認知的発達の遅れです。
最も重篤な病型は、脳全体に影響を及ぼす両側性全身性多小脳回です。この病態は、重度の認知障害、運動障害、そして発作(薬物療法では制御が困難または不可能な持続性強直間代発作)を引き起こします。[ 12 ]
診断 多精子症
脳の多小脳回症の診断は、症状と、遺伝子検査やさまざまな画像技術を含む神経学的検査の結果に基づいて決定されます。
今日では、最も有益な機器診断法は脳の磁気共鳴画像法(MRI)を使用すると考えられている。[ 13 ]
脳波検査は脳機能を評価するために使用されます。
差動診断
鑑別診断は、脳回肥大、裂脳症、脳機能の症候群性障害、小児の特発性全般てんかんおよび局所てんかんなど、他の脳の先天異常との鑑別診断によって行われる。[ 14 ]
処理 多精子症
防止
大脳皮質のこの奇形の発生につながる自然発生的な遺伝子変異の割合がかなり高いことを考慮すると、予防は不可能だと考えられています。
予測
多小脳回症の予後はほとんどの場合不良で、患者の87~94%は再発性てんかんを伴い、事実上治癒不可能な状態となります。両眼に異常がある、あるいは片方の脳半球の回旋の半分以上に病変がある多くの小児は、幼少期に死亡します。