
小児の神経芽細胞腫:原因、診断、治療
最後に見直したもの: 04.07.2025

すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
小児腫瘍学において、最も一般的な頭蓋外腫瘍の 1 つは小児の神経芽細胞腫です。これは、神経堤神経芽細胞、つまり交感神経系の胎児(未熟)神経細胞の胎児性悪性腫瘍です。
疫学
国際神経芽腫リスクグループ(INRG)の統計によると、神経芽腫は世界中の小児における腫瘍性疾患全体の約8%を占め、有病率は白血病と脳腫瘍に次いで3番目に高い。
他のデータによると、神経芽腫は乳児がん全体の約28%を占めています。神経芽腫の症例の3分の1以上は1歳未満の乳児で診断されており、平均診断年齢は19~22ヶ月です。診断例の90%以上は2~5歳の乳児(主に男児)に発生し、発症率は2~3歳でピークを迎えます。5歳以上の乳児の症例は10%未満です。
原因 神経芽細胞腫
神経芽腫の原因を研究する中で、研究者らは、小児におけるこの腫瘍は、胚発生期または出生後早期の発達過程における散発的な遺伝子変異によって発生すると結論付けています。しかし、催奇形性環境因子の影響は特定されていないため、これらの遺伝子変異の原因は不明です。
これらの腫瘍は、縦隔、首、腹部、副腎、腎臓、脊椎、骨盤など、どこにでも発生する可能性があります。
稀ではありますが、乳児の神経芽腫は遺伝性変異に関連している場合があります。具体的には、2番染色体上の膜タンパク質CD246遺伝子(細胞間コミュニケーションを担い、神経系の機能に重要な役割を果たすチロシンキナーゼALKの酵素)の変異、および神経細胞の成熟に関与する4番染色体上のタンパク質PHOX2B遺伝子の変異が挙げられます。
神経芽腫は、小児神経線維腫症 1 型、ベックウィズ・ヴィーデマン症候群、高インスリン性低血糖(神経芽腫性膵炎)と関連している場合もあります。
危険因子
今日では、遺伝は小児神経芽腫の発症リスク因子として認識されています。家族歴におけるこの腫瘍の存在、そして子宮内発育中の遺伝子変異に関連する先天異常などが挙げられます。これは特に、複数の臓器に複数の腫瘍が発生する症例に当てはまります。
この腫瘍のリスクを高める外因性要因は研究者によって特定されていません。
病因
神経芽腫の発生機序は、神経堤細胞の分化および成熟の障害によって引き起こされます。神経堤細胞は、ヒト胎児の外胚葉から神経管の縁に形成される両側性の細胞株です。これらの細胞は移動(移動)し、感覚神経細胞、自律神経細胞、神経内分泌細胞、副腎髄質細胞、頭蓋顔面軟骨細胞、骨細胞、色素細胞など、様々な種類の細胞へと分化します。
神経芽腫では、遊走した神経芽細胞は成熟せず、増殖と分裂を続け、腫瘍を形成します。その病因は、以下の遺伝子変異と関連しています。
- 染色体配列の一部が重複しているか、または胚の神経堤細胞内の RBTN1 タンパク質をコードする 11 番染色体上の LMO1 遺伝子のセグメントが重複している。
- ヒト神経幹細胞の増殖を制御するDUF1220タンパク質をコードする染色体1q21.1上のNBPF10遺伝子のコピー数に変化が生じる疾患です。これらの疾患は、この染色体の重複または欠失(DNAの一部が欠失する状態)を引き起こします。
- 腫瘍抑制遺伝子 ATRX(染色体 Xq21.1 上)の変異を伴う;
- 2番染色体上のN-Myc転写因子遺伝子の追加コピー(増幅)の存在を伴います。この遺伝子は、他の遺伝子の活性を制御し、胎児の組織や臓器の形成に必要なタンパク質の形成過程において前駆細胞の増殖を制御する転写因子(DNA結合タンパク質)の1つをコードしています。この遺伝子の増幅は、がん遺伝子へと変化し、細胞周期の乱れ、細胞増殖の増加、そして腫瘍形成を引き起こします。
症状 神経芽細胞腫
神経芽腫の最初の兆候は非特異的であり、食欲不振(および体重減少)、摂食時の疲労、発熱、関節痛などが含まれることがあります。
臨床症状は原発腫瘍の位置と転移の有無(症例の 60 ~ 73% で発生)によって異なります。
原発性神経芽腫は、神経細胞と同様の起源を持つ副腎髄質に局在することが非常に多く見られます。1歳未満の乳幼児では、症例の35~40%で副腎神経芽腫と診断されます。その症状には、腹痛、発熱、体重減少、骨痛、貧血、あるいはペッパー症候群(重度の肝腫大と呼吸窮迫症候群を伴うびまん性肝障害)などがあります。
後腹膜神経芽腫または小児の後腹膜神経芽腫は、成長するにつれて膀胱や腸を圧迫し始め、排尿や排便に問題が生じたり、脚が腫れたりします(男児の場合は陰嚢が腫れます)。
小児の縦隔神経芽腫(縦隔神経芽腫)は、しばしば上大静脈を圧迫し、顔面、首、腕、胸の上部に腫れ(皮膚が青みがかった赤色になり、皮下結節がみられる)を引き起こすことがあります。咳や喘鳴、呼吸困難(息切れ)、嚥下障害(嚥下困難)が現れ、首、鎖骨上、脇の下のリンパ節腫大が認められます。
腫瘍細胞が骨髄に広がると、貧血、血小板減少症、白血球減少症が起こり、出血傾向が高まります。
眼窩周囲への転移では、目の周りにくまやあざが現れます。また、頭痛やめまい、眼球突出(眼球の突出)、そして神経終末の圧迫による眼瞼下垂(眼瞼下垂)や瞳孔の縮小(縮瞳)を引き起こすこともあります。
小児の腹部神経芽腫または腹腔神経芽腫は、腹部に触知可能な閉塞感、腹部膨満、食欲不振、便秘、血圧上昇を引き起こします。腫瘍が脊髄や神経根を圧迫すると、手足のしびれや筋力低下、起立、這うこと、歩くことの困難につながることがあります。骨が侵されると、骨痛が生じることもあります。
腹腔内の腫瘍がリンパ節損傷を伴うステージ3~4の場合、腫瘍細胞が腎実質に入り込み、小児の腎臓に広範囲にわたる神経芽腫が発生し、腎臓機能が損なわれる可能性があります。
ステージ
- ステージ 1 の神経芽腫は、体の 1 つの領域に限局して孤立している原発性腫瘍であり、両側のリンパ節は影響を受けません。
- 神経芽腫ステージ2。ステージ2Aでは、原発腫瘍は1つの領域に限局しているものの、大きく、両側のリンパ節には転移がありません。ステージ2Bでは、腫瘍が存在する側のリンパ節に転移が認められます。
- 神経芽腫ステージ 3: 原発腫瘍が脊髄または体の正中線を越え、リンパ節に片側または両側の転移が見られます。
- 神経芽腫ステージ4:腫瘍が遠隔リンパ節、骨髄、骨、肝臓、または他の臓器に転移しています。ステージ4Sは、1歳未満の小児で、原発腫瘍が局所に留まり、皮膚、肝臓、または骨髄への転移が認められる場合に診断されます。
国際神経芽腫リスクステージングシステム(INRGSS)
INRGSS では、画像診断で確認され、腫瘍の除去がより困難になる可能性があることを示す要因である画像診断定義リスク要因 (IDRF) を使用します。
INRGSS は神経芽腫を 4 つの段階に分類します。
- L1:腫瘍は発生した場所から転移しておらず、重要な構造物にも増殖していません。頸部、胸部、腹部など、体の一部に限局しています。
- L2: 腫瘍は発生した場所から遠くに広がっていません (転移していません) (たとえば、腹部の左側から胸部の左側に成長している可能性があります) が、少なくとも 1 つの IDRF があります。
- M: 腫瘍が体の遠隔部位に転移しています (MS 段階の腫瘍を除く)。
- MS: 18 か月未満の小児に発生する転移性疾患で、がんが皮膚、肝臓、骨髄のみに転移している状態。
合併症とその結果
神経芽腫は、次のような合併症や症状を特徴とします。
- リンパ節、骨髄、肝臓、皮膚、骨への転移。
- 脊髄の圧迫(痛みを引き起こし、麻痺につながる可能性があります)
- 腫瘍随伴症候群の発症(腫瘍から分泌される特定の化学物質、およびその細胞によって発現される抗原ジシアロガングリオシド GD2 の作用による)。これは、急速な不随意眼球運動、協調運動障害、筋肉のけいれん、および下痢として現れます。
- 一次治療完了後に再発する(臨床実践で示されているように、高リスク神経芽腫では症例の 50% で再発が起こります)。
診断 神経芽細胞腫
小児の神経芽腫の疑いがある場合の診断には、診察、臨床検査、画像検査が必要です。
血液検査と尿検査では、カテコールアミン(ノルエピネフリンおよびドーパミン)とホモバニリン酸またはバニリルマンデル酸(これらのホルモンの代謝中に生成される)の検査が行われます。また、血液検査では神経特異的エノラーゼの検査、血清検査では酵素免疫測定(ELISA)、骨髄検査(穿刺吸引法で採取)が行われます。さらに、遺伝子変異の有無を調べるためにDNA検査が行われ、腫瘍組織の細胞形態学的検査のために生検が行われます。
生検サンプルは採取後、検査室に送られ、病理医(がん細胞の特定に特別な訓練を受けた医師)が顕微鏡で観察します。腫瘍が神経芽腫であるかどうかを確認するために、特別な臨床検査も行われることが多いです。
神経芽腫の場合、臨床検査は腫瘍がどのくらい速く成長または広がるか、またどのような治療法が最も効果的かを判断するのにも役立ちます。
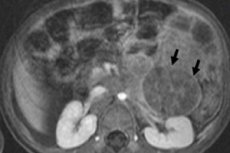
機器診断では、超音波、X線、MRIまたはCT、18F-フルオロデオキシグルコースを導入したPET、またはメタヨードベンジルグアニジンを使用したMIBGスキャン-シンチグラフィーを使用して腫瘍を視覚化します。[ 1 ]
差動診断
鑑別診断には良性神経節腫、神経節芽腫、横紋筋肉腫、腎芽腫が含まれます。
処理 神経芽細胞腫
神経芽腫の治療は、患者のリスクグループ(腫瘍の進行段階)、腫瘍の局在、腫瘍細胞のゲノム特性、そして患児の年齢によって異なります。治療には、経過観察、手術、化学療法、放射線療法、免疫療法、造血幹細胞移植などが含まれる場合があります。
小児神経芽腫に対する術前化学療法または術後化学療法(術前または術後)は、他のがん化学療法と同様に、コース単位で実施されます。薬剤は数日間連続して投与され、その後、体の回復のために休薬期間が設けられます。このサイクルは通常、3~4週間ごとに繰り返されます。
以下の薬剤(およびそれらの組み合わせ)が使用されます:シクロホスファミド、シスプラチンまたはカルボプラチン、ドキソルビシン(アドリアマイシン)、ビンクリスチン、エトポシド。
化学療法薬の一般的な副作用には、脱毛、食欲不振、疲労、吐き気、嘔吐、口内炎、下痢、便秘などがあります。化学療法は骨髄を損傷し、血球数の減少を引き起こす可能性があります。
標的免疫療法(腫瘍抗原GD2を標的とする)では、モノクローナル抗体(抗GD2モノクローナル抗体)であるジヌツキシマブ(ユニツキシン)およびナキシタマブといった薬剤を使用します。これらの薬剤は、顆粒球マクロファージコロニー刺激因子(サイトカインGM-CSF)およびインターロイキン-2と組み合わせて、長期間の点滴静注により静脈内に投与されます。
これらの薬の副作用には、痛み(多くの場合非常に重度)、血圧の低下、心拍数の増加、息切れ(気道の腫れを伴う可能性あり)、体温の上昇、吐き気、嘔吐、下痢、血液の細胞およびミネラル組成の変化などがあります。
高用量化学療法と幹細胞移植後の癌再発リスクを低減するために、高リスク神経芽腫の小児患者は全身性レチノイド、13-シス-レチノイン酸(イソトレチノイン)による治療が行われる。[ 2 ]
神経芽腫の外科的治療 - 腫瘍の切除(例えば、開腹副腎摘出術または腹腔鏡下副腎神経芽腫切除術)、リンパ節郭清(影響を受けたリンパ節の切除)など [ 3 ]
高リスク神経芽腫の場合、放射線療法が用いられることがある。[ 4 ]
防止
小児神経芽腫の原因を考慮すると、妊娠を計画する際に遺伝カウンセリングを受けることが唯一の予防策となるかもしれません。しかし、この腫瘍が遺伝性変異と関連する症例はわずか1~2%であることを念頭に置く必要があります。
予測
乳児神経芽腫は自然に退縮する能力を持っています。
予後マーカー
- 高リスク腫瘍、およびあらゆる年齢層およびあらゆるステージ(ステージ 4S を除く)の小児における神経芽腫(N-MYC 遺伝子の発現増加および N-Myc 癌遺伝子の増幅を伴う)は、予後が不良であり、平均余命に影響を及ぼします。
- 1番染色体または11番染色体の特定の部分が欠損している腫瘍細胞(1p欠失または11q欠失)は、予後不良です。17番染色体の過剰部分(17q増加)も予後不良と関連しています。
- 大量の DNA を持つ神経芽腫細胞は、特に 2 歳未満の小児の場合、予後が良好です。
- 神経栄養因子受容体、特に神経成長因子受容体TrkAを多く持つ神経芽腫は予後が良好です。
小児腫瘍学グループ(COG)リスクグループによる生存率
- 低リスクグループ: 低リスクグループの子供の 5 年生存率は 95% を超えます。
- 中リスクグループ: 中リスクグループの子供の 5 年生存率は 90% ~ 95% です。
- 高リスクグループ: 高リスクグループの子供の 5 年生存率は約 50% です。
小児がんによる死亡の約15%は神経芽腫によるものです。この高リスク悪性腫瘍の長期生存率は40%以下です。全体の5年生存率は67~74%で、1~4歳では43%、生後1年以内に診断された神経芽腫では80%を超えます。
Использованная литература