
下垂体性貧血(小人症)
最後に見直したもの: 12.07.2025

すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
 [
[ 原因 小人症
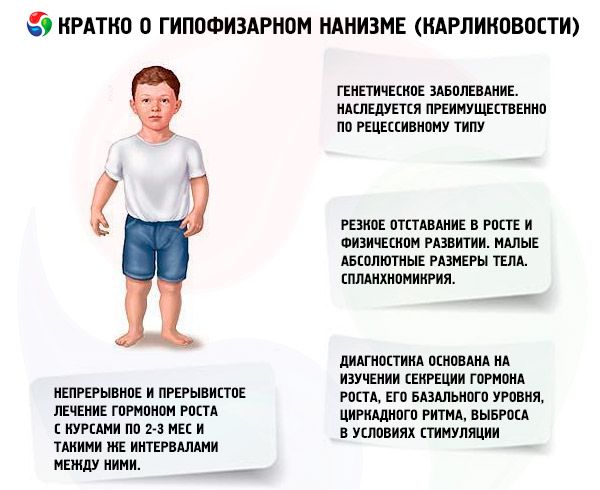
下垂体性小人症のほとんどの形態は遺伝性疾患です。最も一般的なのは汎下垂体機能低下性小人症で、主に劣性遺伝します。この病態の伝達には、常染色体性伝達とX染色体による伝達の2種類があると考えられています。この形態の小人症では、成長ホルモン分泌の欠陥に加えて、ゴナドトロピンと甲状腺刺激ホルモンの分泌が阻害されることが多く見られます。ACTH分泌の阻害は頻度が低く、程度も軽度です。膵ポリペプチドに類似した合成成長ホルモン放出ホルモン(29、40、および44個のアミノ酸残基からなる)などの放出ホルモンを用いた機能研究により、これらの患者のほとんどが視床下部レベルの病変を有し、下垂体前葉の機能不全は二次的なものであることが明らかになっています。下垂体自体の原発性病変はそれほど一般的ではありません。
成長ホルモン単独欠損症を伴う遺伝性小人症は、生物学的活性および感受性の低下を伴い、ロシアおよび近隣諸国で散発的にみられます。この疾患は、アメリカ大陸、近東・中東諸国、そしてアフリカでより多くみられます。血中成長ホルモン含有量、外因性成長ホルモンに対する患者の感受性、免疫反応性インスリン(IRI)、インスリン様成長因子(IGF)I型(ソマトメジンC)およびII型、そして成長ホルモン製剤による治療に対するIGF-1の反応に関する研究結果に基づき、臨床的に類似したタイプの小人症の様々な亜型が特定されています。
最近、IRF-1 および IRF-II の欠乏によって引き起こされるラロン小人症の病因が解明されたほか、前者の欠乏に関連するアフリカのピグミーの小人症の病因も解明されました。
1984 年に、成長ホルモンおよび IGF-1 レベルが高い偽下垂体性小人症の新しい変異型が記述されました。小人症の発生は受容体の欠陥によって説明され、皮膚線維芽細胞の IGF-1 への結合が急激に減少することで証明されています。
現代の状況では、小規模な家族が存在するため、この疾患の多くの孤立した(「特発性」、散発性)症例も遺伝性である可能性があることを強調する必要があります。
350例の症例を分析した結果、小人症の原因が不明な患者は228名(65.2%)でした。このグループには、小人症を繰り返し発症する57家族の患者(1家族あたり2~4例)が含まれており、全患者の28%を占めていました。原因不明(主に遺伝性)の小人症家族の77%で、Rh因子欠損の遺伝との明確な関連性が認められました。小人症患者の家族におけるRh因子の分布は、母親と胎児のRh衝突で観察される分布とは異なり、原則として新生児の溶血性疾患を伴いません(父親がRh陰性で、両親のRh因子がヘテロ接合性の場合は子供など)。成長ホルモン(または成長ホルモン放出ホルモン)の合成を担う遺伝子の活性とRh因子を決定する遺伝子の活性との間に関連があると推測することは可能です。特に、ほとんどのタイプの小人症およびRh因子の欠如は常染色体劣性形質であるためです。しかし、このことは、人口におけるRh陰性者の頻度と比較して小人症が相対的にまれである理由を説明できません。おそらく、まだ知られていない他の要因が重要であると考えられますが、家族性および散発性小人症患者の家系におけるRh因子の分布の特徴は偶然ではない可能性が高いです。
小人症(原発性脳性、脳下垂体性)の患者の多くは、胎児期または幼少期に発症した様々な種類の中枢神経系の器質的病変を有する患者です。この病変を引き起こす解剖学的基質としては、下垂体の発育不全または欠損、トルコ鞍形成異常、下垂体の嚢胞性変性、腫瘍(頭蓋咽頭腫、好色性腺腫、髄膜腫、神経膠腫)による圧迫による萎縮などが挙げられます。小人症は、視床下部-下垂体領域の外傷(子宮内、出産時、または産後)によって引き起こされることがあります。これは、多胎妊娠、骨盤位、足位、または脚を回転させる横向き出産(小人症患者の3分の1以上でみられる出産メカニズム)でよく発生します。感染および毒性による損傷も重要です(子宮内ウイルス感染症、結核、梅毒、マラリア、トキソプラズマ症、幼児期の疾患、新生児敗血症、髄膜炎およびクモ膜脳炎など)。これらのプロセスは、下垂体自体、およびその機能を制御する視床下部中枢に損傷を与え、中枢神経系の正常な機能的接続を阻害する可能性があります。
子宮内胎児病変により、成長ホルモンの分泌は正常であるものの「出生時からの小人症」(大脳原始性小人症、小頭症、体の片側非対称と高ゴナドトロピンを伴うシルバー・ラッセル小人症など)の患者が誕生することがあります。
小人症の身体的発達の障害を悪化させるさらなる要因としては、栄養不足、必須成分(タンパク質欠乏)と微量元素(亜鉛欠乏)の点での不均衡、好ましくない環境条件、および高窒素血症が肝臓受容体の活動に影響を与えたり、肝細胞の代謝に直接影響を与えてソマトメジンの合成を減少させる糸球体腎炎や、ソマトメジンの形成が損なわれた肝硬変などのさまざまな慢性疾患が挙げられます。
病因
下垂体性小人症の患者の多くでは、変化は成長ホルモン分泌の病理やそれに対する感受性に限定されず、下垂体の他の成長ホルモンにも及び、それが内分泌障害と代謝障害のさまざまな組み合わせを引き起こします。
成長ホルモン単独欠乏症における下垂体の形態学的変化については、十分な研究がなされていない。研究対象となった症例では、病理学的異常(頭蓋咽頭腫または頭蓋骨の骨増殖症)が稀に認められた。このタイプの小人症では、ペプチド細胞の先天性発育不全または視床下部の神経伝達物質系の欠陥が観察されることがある。このような症例では、小人症に視神経の形成不全または低形成が併発することがある。視床下部内嚢胞、下垂体腫瘍、および視床下部腫瘍はSTH欠乏症を引き起こし、下垂体組織、特に成長ホルモン産生細胞の圧迫を引き起こす。
小人症は、主に皮質層に起因する骨の菲薄化、骨格の分化および骨化の遅延を特徴とする。内臓は低形成で、時には萎縮し、筋肉の発達も不良である。
症状 小人症
下垂体性小人症の主な症状は、成長と身体発達の著しい遅れです。患者は正常な体重と身長で生まれますが、2~4歳頃から成長が遅れ始めます。
ナニズムに対する積極的治療が登場する以前は、女性では身長120cm未満、男性では身長130cm未満の人が小人症とされていました。現在では、小人の身長は、特定の性別、年齢、または集団における平均標準値から少なくとも2~3シグマの偏差で異なります。ガウス分布曲線に基づいて身長をグラフで評価する方法もあります。この場合、身長によって分類された小人は、対応する集団において平均成長標準値からの遅れが最も大きい個体数が最小となるグループに含められます。
下垂体性小人症は、絶対的な体格が小さいだけでなく、年間成長と身体発達のダイナミクスも小さいことが特徴的です。体格は均整が取れていますが、患者の体型は小児期に典型的なものです。皮膚は青白く、しばしば黄色がかった色調で乾燥しており、これは絶対的または相対的な甲状腺機能不全によるものです。また、チアノーゼ(皮膚の「大理石模様」)が見られる場合もあります。治療を受けていない患者では、早期に老けてしわのある皮膚(老人性皮膚症)が現れます。これは、STHの同化作用が不十分で、細胞世代交代が遅いことが原因です。
頭髪は正常、あるいは乾燥して細く、脆い場合があります。まつ毛が長いのが典型的です。二次毛の成長はしばしば見られません。小人症患者の多く(70~75%)では、トルコ鞍の大きさは変化しませんが、トルコ鞍はしばしば「立卵形」の子供のような形状を維持し、「幼少期」の広い後部を持ち、蝶形骨洞の空気化が遅れています。しかしながら、腫瘍の兆候であるトルコ鞍の肥大、トルコ鞍の背景または入口部に石灰化領域が認められる患者(頭蓋咽頭腫、神経感染症の残存症状)、またはトルコ鞍の減少(発育不全の兆候、下垂体の小型化)がみられる患者もいます。頭蓋内圧亢進症の症状として、頭蓋骨の菲薄化、血管パターンの増加、指圧痕の存在などが観察されます。下垂体性小人症の最も重要な徴候は、骨格の分化と骨化の時期の遅れです。歯系の特徴も骨格の分化と密接に関連しており、乳歯の生え変わりの遅れが見られます。骨格系の発達の最も大きな遅れは、性機能不全と甲状腺機能低下症を伴う小人症患者に観察されます。
ほとんどの患者では性器の発達が著しく遅れていますが、奇形はまれです。男性患者の5.8%に停留精巣が認められました。性機能不全は、二次性徴の発達不全、性欲減退、月経不順を伴います。正常な自発的な性発達は、成長ホルモン単独欠損症の患者と脳性小人症の一部の患者でのみ認められます。
甲状腺機能不全は、小人症のかなり一般的な症状です。甲状腺機能低下症の外部症状は、必ずしも甲状腺の真の機能状態を反映するわけではないことに注意する必要があります。これは、チロキシン(T 4)からトリヨードチロニン(T 3 )への移行の阻害と、ソマトトロピン機能不全の特徴である不活性(可逆性)T 3の形成による相対的甲状腺機能低下症によるものです。
下垂体性小人症における副腎皮質刺激機能の低下は、性機能や甲状腺刺激機能の低下に比べると頻度も程度も低く、ほとんどの患者では特別な矯正は必要ありません。
ほとんどの場合、知能は低下しません。精神的幼児症のような感情的変化がみられます。また、知的障害のない高齢患者では、反応性神経症が観察されることもあります。
有機的な脳病変、特に腫瘍の性質を持つ病変では、尿崩症、両耳側半盲、知的障害などの症状を伴って小人症が発生することがあります。
中枢神経系の器質性症状のない患者の脳の生体電気活動の発達に関する研究では、脳波が未熟さ、高い「幼稚な」脳波電圧の長期にわたる維持、アルファ波の振幅と周波数の不均一性、特に前頭葉および中枢誘導における遅いθ波およびδ波の含有量の急激な増加、過換気に対する明らかな反応、光刺激のリズムに追随する脳波リズムの範囲の低周波数へのシフト(脳神経構造の機能的可動性の低下の証拠)といった特徴を呈することが示されました。高齢患者における脳の電気活動の未熟さは、性的発達不全に起因すること、また、あらゆる年齢層の患者において甲状腺機能低下症が認められることが明らかになりました。
小人症患者の炭水化物代謝は、空腹時血糖値の低下傾向、運動中の血糖値の上昇、内因性インスリンの不足、外因性インスリンに対する感受性の亢進、そして低血糖状態の頻発を特徴とします。後者は主に、患者の体内の抗インスリンホルモンの不足によって説明されます。
内臓は内臓容積の減少、すなわちサイズの縮小を示す。小人症に特有の内臓の機能的変化は報告されていない。収縮期血圧と拡張期血圧の低下、および脈拍振幅の減少を伴う動脈性低血圧がしばしば観察される。心音は抑制され、心筋の栄養変化と自律神経障害により、様々な話題の機能性雑音が聴取される。心電図は低電圧(特に甲状腺機能低下症がある場合)、洞性徐脈または徐脈性不整脈を特徴とし、心電図は音振幅の減少、付加音、および機能性雑音を示す。酸素測定データは、低酸素血症(初期および運動中)、および酸素負債を示す。高齢患者は高血圧を発症することがある。
診断 小人症
小人症の診断と鑑別診断は、病歴データと包括的な臨床検査、放射線学的検査、臨床検査、ホルモン検査に基づいて行われます。患者の成長を評価するために、絶対的な体格に加えて、患者の身長と性別および年齢における平均標準値との差である「成長不足」、患者の身長が特定の基準を満たしているかどうかである「成長年齢」、そして標準化された偏差の指標である「成長年齢」が決定されます。
I = M - Mcp / δ、ここでMは患者の身長、Mcpは特定の性別と年齢における平均正常身長、δはMcpからの二乗偏差です。Iが3未満の場合、小人症の典型例であり、Iが3を超える場合、巨人症の典型例です。この指標は、発達のダイナミクスを評価するために使用できます。
小人症患者のX線検査では、頭蓋内圧亢進、神経感染の残存、石灰化、頭蓋骨癒合症などの兆候が明らかになります。トルコ鞍の大きさ、形状、構造の検査は、下垂体の大きさを特徴付ける間接的な指標と考えられています。病的な成長遅延の最も重要な兆候の一つは、骨格分化の障害です。骨格成熟度を評価するために、骨組織の分化に対応する骨(X線画像)年齢を測定します。骨化不全とは、標準値(年数)からの骨化の遅れの程度であり、骨化係数とは、骨年齢を暦やその他のパラメータで割った商です。
現代の小人症の診断は、成長ホルモンの分泌、その基礎レベル、概日リズム、そして刺激による放出を研究しなければ不可能です。下垂体性小人症の患者の多くは、血清中の成長ホルモン含有量の減少を特徴としています。放射免疫学的方法による測定では、(様々な著者によると)0.87±0.09~1.50±0.64 ng/mlの範囲で、平均基準値は3.81±0.29 ng/mlです。成長ホルモン分泌の概日リズムに関する研究では、健康な人における成長ホルモン含有量は、睡眠の最初の2時間と午前4時から6時に最大になることが示されています。小人症においても、これらの時間帯における成長ホルモン含有量が減少しています。
成長ホルモン機能の予備能を調べるために、様々な刺激剤を用い、投与前後の成長ホルモン含有量を調べます。検査のための採血は2~3時間、30分ごとに行います。刺激後の成長ホルモン放出量は、少なくとも7~10 ng/mlであれば正常とみなされますが、20~40 ng/mlに達することもあります。いずれかのサンプルで反応が見られない場合は、別の刺激剤を用いて再検査を行います。2~3つの異なるサンプルで成長ホルモン放出が見られない場合、成長ホルモン不足が証明されたとみなされます。
最も一般的に用いられる刺激試験は、患者の体重1kgあたり0.1単位(0.75~1.5単位)のインスリンを静脈内投与し、低血糖(初期血糖値と比較して50%の低下)を達成した時点で、上記の方法に従って血清中の成長ホルモン濃度を測定するものです。重度の低血糖が発現した場合は、試験を中断し、患者にブドウ糖を静脈内投与します。これは最も一般的で古典的な診断方法です。
TRHを200~500マイクログラム静脈内投与します。ホルモンの予備量を効果的に特定し、合併症を引き起こしません。インスリン検査と併用することで、視床下部-下垂体系の損傷レベルを判定できます。TRHに陽性反応を示し、インスリン低血糖反応がない場合は、下垂体は正常であり、視床下部レベルの損傷があることを示します。TRHと低血糖に陰性反応がある場合は、下垂体自体の損傷があることを示します。
TRH、LH-RH 300 mcg を静脈内投与した場合の投与量は、前のものと同様です。
ヒトSGHは、膵臓腫瘍から単離された生物活性化合物の合成類似体です。現在、合成SGHには、アミノ酸残基数が29、40、44の3種類があります。患者の体重1kgあたり1~3μgの用量で静脈内投与されます。投与後15~20分でSTHの放出が観察され、この検査は内因性成長ホルモンの蓄積を明らかにする上で他の検査よりも効果的です。SGH反応が陽性の場合、視床下部レベルの成長ホルモン機能障害と下垂体機能の健全性を示します。アミノ酸(L-アルギニンモノクロリド、オルニチン、トリプトファン、グリシン、ロイシン)を患者の体重1kgあたり0.25~0.5gの用量で静脈内投与します。SGHの蓄積の研究に有効です。アレルギー反応を引き起こす可能性があります。
L-ドーパを250~500マイクログラム経口投与します。効果があり、患者の忍容性も良好です。
グルカゴン、臭素エルゴクリプチン(パーロデル)、リジンバソプレシン、クロニジン、および一定量の自転車エルゴメータ負荷を用いたテストも使用されます。
成長ホルモン機能の状態の研究は、小人症の診断だけでなく、治療法の正当な選択にも必要です。なぜなら、成長ホルモンによる治療は、内因性成長ホルモンが不足している場合にのみ合理的だからです。
小人症の診断には、インスリン様成長因子、すなわちソマトメジン(特にIGF-1またはソマトメジンC)の含有量を調べることが非常に重要です。これらは組織レベルで成長ホルモンの作用を媒介する物質です。小人症ではソマトメジンCの含有量が正常値より低く、先端巨大症では正常値より高いことが知られています。ラロンが報告した小人症は、STHの産生は正常ですが、IGF-1およびIGF-IIの形成に異常がある疾患の一種です。このような患者を成長ホルモンで治療しても効果はありません。
下垂体のソマトトロピン機能の間接的な指標は、アルカリホスファターゼの活性と血清中の無機リン含有量です。ソマトトロピン機能低下状態では、これらの指標は減少します。汎下垂体機能低下型の小人症では、ゴナドトロピン(多くの場合TSH)の分泌が減少し、これに伴って性腺機能(アンドロゲンまたはエストロゲンの欠乏)、甲状腺機能(T3 、 T4 、タンパク質結合ヨウ素(PBI)のレベルの減少、甲状腺によるヨウ素131の蓄積)、および副腎機能(血漿中のコルチゾールおよび17-OCS量の減少、尿中への17-KCおよび17-OCSの排泄、リンパ球増多)が低下します。
下垂体(視床下部-下垂体)遺伝性小人症のすべてのタイプは、常染色体劣性(より一般的)または常染色体優性遺伝による家族内の子供の再発性疾患、特定の性別、年齢、人口の平均成長基準から少なくとも2~3°の遅れを伴う2~4歳の成長遅延および身体発達、低い自発的な年間成長ダイナミクス、骨化の遅延を特徴とします。成長ホルモンレベルが低い場合(2~3回の刺激試験で7 ng / ml未満)、成長ホルモン療法は非常に効果的です(少なくとも年間7 cmの身長増加をもたらします)。成長ホルモンレベルが正常または高い場合(生物学的に不活性)、ホルモンに対する感受性は維持されます。知能の変化は観察されません。
成長ホルモンに対する組織の不応性を伴う遺伝性小人症は、臨床像は成長ホルモン単独欠損症に類似しますが、成長ホルモン療法は無効です。このグループでは、IRFレベルに応じて、以下の主要な病型に区別されます。正常含量(IRF受容体欠損)および減少型(ラロン型小人症(IRF-1およびIRF-II欠損))、そしてアフリカのピグミーにみられる型(IRF-1欠損)です。
脳性小人症は、子宮内または出産後の中枢神経系の損傷に関連する家族内の孤立した疾患を特徴とし、中枢神経系の明らかな器質的変化を伴い、視覚器官の病理、尿崩症の存在、性腺刺激ホルモン機能の保持、および知能の変化を伴うことが多い。
性腺形成不全症および性腺機能不全症の中には、顕著な低身長を伴うものもあり、特にシェレシェフスキー・ターナー症候群や「ターナー型」(モザイク型)精巣形成不全症候群が顕著です。細胞遺伝学的検査(性染色体、核型)は鑑別診断に役立ち、染色体異常、体細胞および性機能の発達における特徴的な異常、内因性成長ホルモンの正常または高値、成長ホルモン治療への不応性などが明らかになります。
低身長に伴う内分泌疾患の中で、原発性甲状腺機能低下症は特に注目すべき疾患です。これは、甲状腺の先天性低形成または無形成、甲状腺の異形成、甲状腺ホルモン生合成における酵素欠損、甲状腺の早期自己免疫性障害によって引き起こされます。これらの疾患では、血清中のTSH高値、T4およびT3低下といった甲状腺機能低下症の徴候が顕著です。自己免疫性粘液水腫では、血中にチログロブリン、甲状腺組織のミクロソーム分画および核分画に対する抗体が検出され、成長ホルモンレベルは正常または低下しています。臨床効果は、甲状腺機能低下症のみを補うことで得られます。
低身長は、成長帯の早期閉鎖による早熟な性的発達および副腎性器症候群を伴います。イツェンコ・クッシング病は、成長ホルモン分泌に対するグルココルチコイドの阻害作用およびその異化作用により小児期に発症します。モーリアック症候群は、重度のインスリン依存型糖尿病患者の低身長および幼児期の特徴です。
下垂体性小人症は、慢性代謝障害(肝臓、腎臓、消化管の疾患)、慢性低酸素症(心血管系および呼吸器系の疾患、貧血)による体性身体発達遅延、筋骨格系の全身性疾患(軟骨異栄養症、骨形成不全症、骨腫症)などと区別する必要があります。
機能的(体質的)成長遅延は、一見健康な青年期の思春期開始の遅れに伴って時に観察されます。これは主に、一時的なゴナドトロピン活性の低下と関連していることが分かっています。成長ホルモン分泌は通常、障害されないか、わずかに減少します。ゴナドトロピンを刺激することで、性的発達と成長の両方を促進できます。
家族的な性質による低身長は、生理的発達の変異として考慮されるべきである。
どのように調べる?
どのようなテストが必要ですか?
連絡先
処理 小人症
小人症の治療は長期にわたるプロセスです。医師は、2つの基本原則を守りながら、最大の臨床効果を得るために、成長に影響を与える手段を時間をかけて分散させる必要があります。
- 治療によって誘発される発達を生理学的条件に最大限近づけること。
- 骨端線の成長領域を保護します。
小人症の治療における長年の経験から、以下の段階的な治療法が適切であると考えています。成人患者における小人症の診断は、通常、疑義を生じません。小さな小児の場合、臨床像が不明瞭な場合は、ホルモン療法を行わずに6~12ヶ月間の観察期間を設ける診断期間が必要です。この期間中は、総合的な全身強化療法が処方されます。適切な栄養摂取に加え、食事中の動物性タンパク質、野菜、果物、ビタミンA、D、カルシウム、リン製剤の摂取量を増やします。こうした背景を踏まえ、成長と身体発達に十分な変化が見られないこと、そして検査中に内分泌疾患が検出されることが、ホルモン療法開始の根拠となります。
下垂体性小人症の主な病因療法はヒト成長ホルモンの使用です。これは、小人症のほとんどの症例の発症が、間違いなく何らかの形のヒト成長ホルモン欠乏症に依存しているためです。このホルモンの種特異性のため、ヒトおよび霊長類の成長ホルモンのみがヒトに対して有効です。非感染性および非腫瘍性疾患で死亡した人の下垂体から単離された薬が、臨床で広く使用されています。ヒト成長ホルモンは、遺伝子工学により大腸菌を使用して細菌合成によって得られます。ヒト成長ホルモンは化学的にも合成されますが、非常に高価であるため、臨床では実際には使用されていません。内因性成長ホルモンの欠乏が証明され、骨格分化が13〜14歳の典型的なレベルを超えない患者が、成長ホルモン治療の対象となります。治療に年齢制限はありません。
治療初期に使用できる最小有効用量は、体重1kgあたり0.03~0.06mgです。最も効果的な用量は、週3回2~4mgの投与です。単回投与量を10mgに増量しても、成長効果は十分に増加せず、ソマトトロピンに対する抗体が急速に形成されました。
わが国では、1960年からヒト成長ホルモンの研究が行われてきました。2種類の治療計画がテストされています。1つは継続的治療、もう1つは間欠的治療で、2〜3か月のコースで各コース間の間隔は同じです。治療1年目の患者の身長の平均増加は9.52±0.39 cm、体重の増加は4.4±0.14 kgでした。長期の継続的治療では、平均身長の増加は0.82 cm /月、体重は0.38 kg /月でした。間欠的治療では、それぞれ0.75 cm /月と0.4 kg /月でした。継続的治療では身長がより急速に伸びますが、1〜1.5年後には効果が急激に減少します。間欠的治療では、効果が3〜4年間維持されるため、コース治療計画の方が適切であると考えられます。 IGF-I(ソマトメジンC)濃度の測定は、ソマトトロピン薬による治療に対する患者の感受性を測る信頼性の高い指標となります。ソマトトロピン投与後のIGF-I濃度の上昇は、治療効果の予測に役立ちます。ソマトトロピン治療の重要な利点は、ソマトトロピン投与に伴う骨化の促進がないことです。
小人症の治療に最も重要な手段は、タンパク質合成を促進し、内因性成長ホルモンのレベルを上げることで成長を刺激するアナボリックステロイドの使用です。治療は数年間行われ、一部の薬剤を他の薬剤に徐々に置き換え、活性の低い化合物から活性の高い化合物へと切り替えていきます。アナボリック薬の変更は、2〜3年後に成長効果が低下し、成長がさらに促進された場合に示されます。治療はコースで行われます(休薬期間は治療期間の半分である必要があります)。依存症の場合は、より長い休止期間も示されます(最大4〜6か月)。一度に処方されるアナボリックステロイドは1つだけです。2つ以上の薬剤を併用することは、代謝効果と成長効果を高めないため不適切です。後者は、主に患者の年齢と治療開始時の骨格の分化の程度によって異なります。最良の効果は、骨格骨化が14歳の特徴的なレベルを超えない16〜18歳未満の患者で観察されます。診断後すぐに、通常は5〜7歳から治療を開始することをお勧めします。治療前には、成長を刺激すると同時に骨格の分化を促進するゴナドトロピンと性ホルモンの処方を避ける必要があります。アナボリックステロイドの投与量の原則は、最小有効量から徐々に増やすことです。最も一般的な薬の推奨用量:ネロボル(メタンドロステノール、ダイアナボル)-経口で体重1kgあたり0.1〜0.15mg /日。ネロボリル(デュラボリン)-筋肉内に体重1kgあたり1mg /月、毎月の用量は、それぞれ15日または10日後に2〜3回投与されます。レタボリル(デカデュラボリン)-体重1kgあたり1mgを月に1回筋肉内投与します。指定された用量を超えると、アンドロゲン化につながる可能性があります。生理学的用量では、これらの化合物は性器の状態や骨格の分化に大きな影響を与えないため、男女を問わず長期間使用できます。過剰摂取または個人の感受性の増加の場合、一部の患者は男性化の兆候を発現する可能性があり、治療を中止するとすぐに退行するため、女の子は婦人科医の監督下にある必要があります。17番目の位置でメチル化またはエチル化された経口薬は、胆汁うっ滞効果を引き起こすことがあるため、肝疾患の場合は、非経口同化化合物を優先するか、経口薬を胆汁分泌促進剤と併用する必要があります。ごくまれに、同化ステロイドによる治療がアレルギー反応(かゆみ、発疹)を引き起こすことがあります。合併症がない場合、アナボリックステロイドは成長効果が認められる限り(最長16~18歳、場合によってはそれ以上)使用されます。治療は、一般的な筋力強化療法を背景に行われます。
患者に甲状腺機能低下の兆候がある場合は、甲状腺薬(チロキシン、チロイジン、チロトム)が個別に選択された用量で同時に処方されます。
男児の治療では、次のステップはヒト絨毛性ゴナドトロピンの投与です。この薬剤は15~16歳以降に使用され、ライディッヒ細胞を刺激することで性的発達と成長(自身のアンドロゲンの同化作用による)を促進するため、しばしばそれよりも高い年齢で使用されます。1000~1500 IUの用量を週1~2回、2ヶ月間のコースで筋肉内投与し、年間2~3回を超えて投与しないでください。効果が不十分な場合は、16歳以上の男児に対するヒト絨毛性ゴナドトロピンによる治療と、少量のアンドロゲン(メチルテストステロンを舌下投与で1日5~10mg)の投与を交互に行います。
16歳以上の女子は、通常の性周期を模倣した少量のエストロゲン投与から治療を開始できます。治療は毎月3週間行い、その後休薬します。周期の第2期である3週目からは、絨毛性ゴナドトロピンを1000~1500 IUの用量で週3~5回投与するか、黄体ホルモン作用を持つ薬剤(プレグニン、プロゲステロン)を処方します。
治療の最終段階(成長帯の閉鎖後)は、患者の性別に応じた治療量の性ホルモンを持続的に投与することです。これにより、性器の完全な発達、二次性徴の発現、性欲および性的能力の確保が促進されます。女性患者の治療には、エストロゲン・プロゲストーゲン配合剤(ノンオブロン、ビセクリン、インフェクンジン、リゲビドン)が、男性患者の治療には、徐放性アンドロゲン製剤(テストネート、サスタノン250、オムナドレン250)が適しています。
総合的な強化療法(レジメン、タンパク質・植物性食事療法、ビタミン療法、バイオスティミュラント)を実施します。亜鉛製剤の使用が適応となります。その作用機序は、IGF-1(インスリン様成長因子I)の活性を高めることが主な役割です。
中枢神経系の器質性病変が存在する場合、抗炎症療法、骨吸収療法、脱水療法が行われます。標的を絞った体系的な治療は、良好な効果をもたらします。長期にわたる段階的な治療の結果、男女ともに175名の小人症患者のうち、148名(80.4%)が130cm以上、92名(52.5%)が140cm以上、32名(18.3%)が150~160cm以上の身長を達成しました。同時に、37名(21.2%)は30cm、107名(61.1%)は31~50cm、31名(17.7%)は51~60cm以上の身長増加を達成しました。
予測
予後は小人症の形態によって異なります。遺伝性の小人症の場合、生命予後は良好です。下垂体腫瘍および中枢神経系の器質性損傷を伴う場合、予後は主要な病理学的過程の発達のダイナミクスによって決定されます。現代の治療法は、患者の身体能力と労働能力を大幅に向上させ、平均余命を延ばしました。積極的治療期間中は、患者は2~3ヶ月ごとに医師の診察を受け、維持療法は6~12ヶ月ごとに受ける必要があります。
患者の知的・身体的能力に応じた雇用は、患者の社会適応にとって極めて重要です。
肉体的に大きな負担を伴わず、知的能力、精密な作業を行う能力、言語を発揮できる職業を選択することをお勧めします。