
プリオン-プリオン病の原因物質
最後に見直したもの: 06.07.2025

すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
緩やかなウイルス感染は特別な基準によって特徴付けられます:
- 異常に長い潜伏期間(数か月、数年)
- 臓器や組織、特に中枢神経系の特定の病変。
- 病気のゆっくりとした着実な進行。
- 避けられない致命的な結果。

急性ウイルス感染症を引き起こす病原体の中には、緩徐なウイルス感染症を引き起こすものもあります。例えば、麻疹ウイルスはSSPEを引き起こすことがあり、風疹ウイルスは進行性先天性風疹および風疹全脳炎を引き起こします。
動物における典型的な緩徐ウイルス感染症は、レトロウイルスであるビスナ/マディウイルスによって引き起こされます。これは、羊における緩徐ウイルス感染症と進行性肺炎の原因物質です。脳の白質が破壊され、麻痺(ビスナ:萎縮)が生じ、肺と脾臓に慢性的な炎症が生じます。
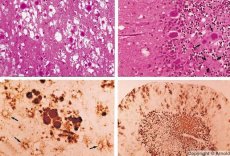
スローウイルス感染症と特徴的に類似した疾患は、プリオンによって引き起こされます。プリオンは、プリオン感染症の原因物質です。プリオン病は、ヒトおよび動物の中枢神経系の進行性疾患群です。ヒトでは、中枢神経系の機能障害、人格変化、運動障害などがみられます。この疾患の症状は通常数ヶ月から数年続き、最終的には死に至ります。かつて、プリオン感染症は、いわゆるスローウイルス感染症の原因物質と一括して考えられていました。
プリオン病を引き起こす病原体の中には、まずリンパ組織に蓄積するものがいくつかあります。脳内に侵入したプリオンは大量に蓄積し、アミロイドーシス(細胞外タンパク質異常症、アミロイドの沈着と組織の萎縮および硬化の進行を特徴とする)およびアストロサイトーシス(アストロサイトの神経膠細胞の増殖、グリア線維の過剰産生)を引き起こします。脳内には、タンパク質またはアミロイドの凝集体である原線維と海綿状の変化(伝達性海綿状脳症)が形成されます。その結果、行動の変化、運動協調の障害、致命的な結果を伴う衰弱が生じます。免疫は形成されません。プリオン病は、体の正常な機能に必要な細胞タンパク質の誤った折り畳み(正しい構造の破壊)の結果として発症する構造疾患です。プリオンの感染経路は多岐にわたります。
- 消化経路 - 動物由来の感染製品、生の牛の臓器からの食品添加物など:
- 輸血、動物由来の薬剤の投与、臓器および組織の移植、感染した外科用および歯科用器具の使用による感染。
- 免疫生物学的製剤による伝染(病気の羊から採取した脳ホルモールワクチンにより 1500 頭の羊が PrP''' に感染したことが知られている)。
病的プリオンは腸管に侵入すると、血液およびリンパ液中に輸送されます。脾臓、虫垂、扁桃腺などのリンパ組織で末梢複製された後、末梢神経を介して脳に移行します(神経侵襲)。血液脳関門を通過してプリオンが脳に直接侵入する可能性もあります。以前は、病的プリオンが蓄積する組織は中枢神経系のみであると考えられていましたが、この仮説を覆す研究が発表されました。脾臓におけるプリオンの蓄積は、濾胞樹状細胞の増加と機能に関連していることが判明しました。
 [
[ プリオンの特性
分子量33~35 kDaのプリオンタンパク質の正常な細胞アイソフォームは、プリオンタンパク質遺伝子(プリオン遺伝子 - PrNPはヒト20番染色体上に存在する)によって決定されます。この正常な遺伝子は細胞表面に発現し(分子の糖タンパク質によって膜に固定されています)、プロテアーゼに感受性があります。神経インパルスの伝達、日周期、酸化プロセスを制御し、中枢神経系における銅代謝および骨髄幹細胞の分裂制御に関与しています。さらに、プリオン遺伝子は脾臓、リンパ節、皮膚、消化管、濾胞樹状細胞にも存在します。
病的プリオンの増殖
プリオンが変異体へと変化する過程は、両者間の速度論的に制御された平衡が破綻した際に起こります。この過程は、病的プリオン(PrP)または外因性プリオンの増加によって促進されます。PrPは細胞膜に固定された正常なタンパク質です。PrP'は球状の疎水性タンパク質で、細胞表面で自身とPrP''と凝集体を形成します。その結果、PrP'はPrP''へと変化し、このサイクルが継続されます。病的プリオン(PrP''')はニューロンに蓄積し、細胞をスポンジ状の外観にします。
クールー
プリオン病は、ニューギニア島東部のパプア人(震え、震えを意味する)の間でかつてよく見られた病気です。この病気の感染性はK.ガジュセクによって証明されました。病原体は、プリオンに感染した死者の脳を加熱不十分に食べるという儀式的な人食いによって食物を介して伝染します。中枢神経系が損傷し、運動機能や歩行能力が低下し、悪寒や多幸感(「笑い死」)が現れます。潜伏期間は5~30年で、患者は1年後に死亡します。
クロイツフェルト・ヤコブ病
プリオン病は、認知症、視覚障害、小脳障害、運動障害などの症状を呈し、古典的なクロイツフェルト・ヤコブ病では発症後4~5ヶ月、新しいクロイツフェルト・ヤコブ病では発症後3~14ヶ月で致命的となる。潜伏期間は20年に達することもある。感染経路や原因は様々である。
- 牛海綿状脳症に罹患した牛の肉や脳など、加熱処理が不十分な動物性食品を摂取する場合。
- 角膜移植などの組織移植、輸血、動物由来のホルモンやその他の生理活性物質の使用、腸線の使用、汚染された、または十分に滅菌されていない手術器具、切開操作中。
- PrR の過剰産生や、PrR' を PrR に変換するプロセスを刺激するその他の状態の場合」
この疾患は、プリオン遺伝子領域の変異または挿入によっても発症することがあります。クロイツフェルト・ヤコブ病の遺伝的素因があるため、家族性疾患であることは一般的です。新しいタイプのクロイツフェルト・ヤコブ病では、典型的なタイプ(平均年齢65歳)と比較して、より若い年齢(平均年齢28歳)で発症します。新しいタイプのクロイツフェルト・ヤコブ病では、異常なプリオンタンパク質が中枢神経系だけでなく、扁桃腺を含むリンパ網様組織にも蓄積します。
ゲルストマン・シュトロイスラー・シャインカー症候群
遺伝性プリオン病は、認知症、筋緊張低下、嚥下障害(嚥下困難)、構音障害を伴います。家族性であることが多いです。潜伏期は5~30年です。発症は50~60歳で、病状は5~13年です。
遺伝性致死性不眠症
進行性の不眠症、交感神経過敏症(高血圧、高体温、多汗症、頻脈)、振戦、運動失調、多クローン性幻覚を伴う自己免疫疾患。睡眠は著しく阻害され、心血管不全が進行すると死に至る。
削る
スクレイピー(英語の scrape - 削る)は、羊や山羊のプリオン病(疥癬)であり、中枢神経系の損傷、進行性の運動障害、重度の皮膚のかゆみ(疥癬)を伴って発生し、最終的には動物の死に至ります。
牛海綿状脳症
牛の病気で、中枢神経系の損傷、運動協調障害、そして動物の避けられない死を特徴とします。この病気の流行はイギリスで初めて発生しました。病原性プリオンを含む肉骨粉を動物に給餌したことが原因とされています。潜伏期間は1.5年から15年です。動物の脳、脊髄、眼球が最も感染しやすい部位です。