
シャルコー・マリー・トゥース病。
最後に見直したもの: 12.07.2025

すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
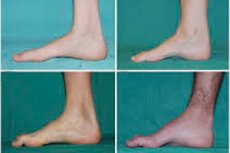
腓骨筋萎縮症、シャルコー・マリー・トゥース症候群または病は、末梢神経に損傷を伴う慢性の遺伝性疾患のグループです。
ICD-10によると、神経系疾患の項では、この疾患のコードはG60.0(遺伝性運動感覚神経障害)です。また、希少疾患のリストにも含まれています。
疫学
臨床統計によれば、シャルコー・マリー・トゥース病のすべてのタイプの有病率は人口 10 万人あたり 19 例です (他の情報源によると、人口 2.5-10 千人あたり 1 例)。
CMTタイプ1は症例の約3分の2(人口5,000~7,000人に1人)を占め、その約70%はPMP22遺伝子の重複に関連しています。世界中で120万人以上がこのタイプの疾患に苦しんでいます。
CMTタイプ4の発生率は10,000人の子供あたり1~5例と推定されています。[ 1 ]
原因 シャルコー・マリー・トゥース病
多発神経障害症候群の分類によると、腓骨筋萎縮症、シャルコー・マリー・トゥース神経性筋萎縮症、またはシャルコー・マリー・トゥース病(CMTと略される)は、遺伝的に決定される運動感覚性多発神経障害を指します。[ 2 ]
つまり、その発症原因は遺伝子変異です。そして、遺伝子異常の性質に応じて、この症候群は脱髄型と軸索型に大きく分けられます。前者のグループには、シャルコー・マリー・トゥース病1型(CMT1)が含まれます。これは、膜貫通型末梢ミエリンタンパク質22をコードする17番染色体上のPMP22遺伝子の重複によって発症します。その結果、軸索鞘(神経細胞突起)の分節的な脱髄と神経信号伝導速度の低下が起こります。さらに、他の遺伝子の変異も発生する可能性があります。
軸索型はシャルコー・マリー・トゥース病2型(CMT2)であり、軸索自体に影響を与え、ミトコンドリア融合と末梢神経細胞内の機能的なミトコンドリアネットワークの形成に不可欠な膜タンパク質ミトフシン-2をコードするMFN2遺伝子(座位1p36.22)の病理学的変化と関連しています。CMT2には、特定の遺伝子の変異を伴う12種類以上のサブタイプが存在します。
現在、100種類以上の遺伝子が特定されており、その損傷は遺伝によって伝達され、シャルコー・マリー・トゥース病の様々なサブタイプを引き起こします。例えば、RAB7遺伝子の変異はCMTタイプ2Bを引き起こし、SH3TC2遺伝子(シュワン細胞の膜タンパク質の一つをコードする遺伝子)の変異はCMTタイプ4Cを引き起こします。CMTタイプ4Cは小児期に発症し、運動ニューロンと感覚ニューロンの脱髄を特徴とします(この疾患のタイプ4には15の病型があります)。
まれなタイプ 3 CMT (デジェリン・ソッタス症候群と呼ばれる) は幼少期に発症し始め、PMP22、MPZ、EGR2 などの遺伝子の変異によって引き起こされます。
CMTタイプ5が5〜12歳で発症すると、運動神経障害(下肢の痙性麻痺の形態)が見られるだけでなく、視神経と聴神経の損傷も見られます。
筋力低下、視神経萎縮(視力喪失を伴う)、バランス障害は、CMT タイプ 6 の特徴です。また、タイプ 7 のシャルコー・マリー・トゥース病では、運動感覚神経障害だけでなく、網膜色素変性症という網膜疾患も見られます。
男性に多くみられるX連鎖性CMT、または四肢麻痺(両腕と両脚の運動麻痺)を伴うシャルコー・マリー・トゥース病は脱髄型で、X染色体長腕のGJB1遺伝子の変異に起因すると考えられています。この遺伝子は、神経信号の伝達を制御するシュワン細胞とオリゴデンドロサイトの膜貫通タンパク質であるコネキシン32をコードしています。[ 3 ]
危険因子
CMT の主な危険因子は、家族歴、つまり近親者におけるこの疾患の病歴です。
遺伝学者によると、両親がシャルコー・マリー・トゥース病の常染色体劣性遺伝子の保因者である場合、この病気を発症する子供が生まれるリスクは25%です。また、子供がこの遺伝子の保因者(ただし症状は現れない)になるリスクは50%と推定されています。
X連鎖遺伝(変異遺伝子が女性のX染色体上にある場合)の場合、母親から息子に遺伝子が受け継がれ、CMTを発症するリスクは50%です。女の子が生まれたときには病気が発症しないかもしれませんが、娘の息子(孫)が欠陥遺伝子を受け継ぎ、病気を発症する可能性があります。
病因
シャルコー・マリー・トゥース病のどのタイプにおいても、その病因は末梢神経(運動神経と感覚神経)の遺伝的異常によって引き起こされます。
CMT 型が脱髄性の場合、末梢神経の軸索を保護する髄鞘の破壊または欠陥により、末梢神経系(脳、筋肉、感覚器官の間)における神経インパルスの伝達が遅くなります。
軸索型の疾患では、軸索自体が影響を受け、神経信号の強度に悪影響を及ぼし、筋肉や感覚器官を十分に刺激するには不十分となります。
こちらもお読みください:
シャルコー・マリー・トゥース症候群はどのように伝染するのでしょうか? 欠陥遺伝子は常染色体優性遺伝または常染色体劣性遺伝で受け継がれます。
最も一般的なタイプである常染色体優性遺伝は、変異遺伝子のコピーが1つ(両親のどちらかが保有)の場合に発生します。そして、生まれた子供にCMTが遺伝する確率は50%と推定されています。[ 4 ]
常染色体劣性遺伝の場合、病気が発症するには欠陥遺伝子のコピー 2 つ (病気の兆候を示さない両親から 1 つずつ) が必要です。
症例の40~50%では常染色体優性遺伝性脱髄(CMTタイプ1)が起こり、12~26%では軸索CMT(タイプ2)が起こります。また、症例の10~15%ではX連鎖遺伝が観察されます。[ 5 ]
症状 シャルコー・マリー・トゥース病
通常、この病気の最初の兆候は幼少期から青年期に現れ始め、生涯を通じて徐々に進行しますが、後になってから症状が現れることもあります。症状の組み合わせは多様であり、病気の進行速度や重症度を予測することは不可能です。
初期段階の典型的な症状としては、全身倦怠感の増加、足、足首、脛の筋肉の緊張低下(筋力低下)、反射神経の低下などが挙げられます。これらの症状により足の動きが困難になり、足が上がりすぎる歩行障害(歩行障害)につながり、つまずいたり転倒したりすることがしばしばあります。シャルコー・マリー・トゥース病の小児における兆候としては、顕著な不器用さや、年齢に似合わない両足下垂を伴う歩行障害などが挙げられます。また、足の変形も特徴的で、ハイアーチ(空洞足)や重度の扁平足、弯曲(ハンマー型)趾などが挙げられます。
筋緊張低下を背景につま先立ちで歩く場合、神経科医は子供が CMT タイプ 4 であると疑う可能性があり、その場合、子供は思春期までに歩行できなくなる可能性があります。
病気が進行するにつれて、筋萎縮と筋力低下が上肢に広がり、微細運動能力や通常の手作業が困難になります。触覚や温冷感の低下、手足のしびれなどは、感覚神経の軸索損傷を示唆します。
小児期に発症するシャルコー・マリー・トゥース病の 3 型および 6 型では、感覚失調 (運動とバランスの協調障害)、筋肉のけいれんと震え、顔面神経の損傷、眼振を伴う視神経萎縮、および難聴が観察されます。
後期には、制御できない震え(振戦)や頻繁な筋肉のけいれんが発生する可能性があり、運動障害により筋肉、関節、神経の痛みが生じる可能性があります。
合併症とその結果
シャルコー・マリー・トゥース病には、次のような合併症や後遺症が現れることがあります。
- 捻挫や骨折の頻度が増える
- 関節周囲の筋肉および腱の短縮に関連する拘縮;
- 脊柱側弯症(脊椎の湾曲)
- 呼吸障害 - 横隔膜の筋肉を支配する神経線維が損傷した場合:
- 自力で移動する能力の喪失。
診断 シャルコー・マリー・トゥース病
診断には、臨床検査、病歴(家族歴を含む)、神経学的検査および全身検査が含まれます。
可動域、感覚、腱反射を確認するための検査が行われます。神経伝導は、機器診断(筋電図検査または電気神経筋図検査)によって評価できます。超音波検査やMRIが必要になる場合もあります。[ 6 ]
血液サンプル中のCMTを引き起こす最も一般的な遺伝子変異を検出するための遺伝子検査またはDNA検査は、現在すべての疾患の種類でDNA検査が利用可能ではないため、限られています。詳細については、遺伝子検査をご覧ください。
場合によっては、末梢神経(通常は腓骨神経)の生検が行われます。
差動診断
鑑別診断には、他の末梢神経障害、デュシェンヌ型筋ジストロフィー、脊髄症および筋無力症候群、糖尿病性神経障害、多発性および筋萎縮性側索硬化症の脊髄症、ギランバレー症候群、腓骨神経の外傷および萎縮(脊椎の腰椎椎間板に挟まれた場合を含む)、小脳または視床の損傷、化学療法の副作用(ビンクリスチンやパクリタキセルなどの細胞増殖抑制剤による治療中)などが含まれる。[ 7 ]
連絡先
処理 シャルコー・マリー・トゥース病
現在、この遺伝性疾患の治療には、運動療法(筋肉の強化とストレッチを目的とします)、作業療法(手の筋力低下の患者を支援します)、そして歩行を容易にするための整形外科用器具の使用が含まれます。必要に応じて、鎮痛剤や抗けいれん剤が処方されます。[ 8 ]
重度の扁平足の場合は骨切り術が行われ、かかとの変形の場合には外科的矯正(関節固定術)が適応となります。[ 9 ]
疾患の遺伝的要素と治療法の両方について研究が進められています。幹細胞、特定のホルモン、レシチン、アスコルビン酸などの使用は、まだ良い結果をもたらしていません。
しかし、近年の研究のおかげで、シャルコー・マリー・トゥース病の治療に新たな可能性が近い将来に現れるかもしれません。フランスの企業Pharnextは2014年から、成人のCMTタイプ1の治療薬PXT3003の開発に取り組んでおり、2019年半ばからは臨床試験が進行中です。PXT3003は、PMP22遺伝子の発現増加を抑制し、末梢神経の髄鞘形成を改善し、神経筋症状を緩和します。
医療企業Sarepta Therapeutics(米国)の専門家たちは、1型シャルコー・マリー・トゥース病の遺伝子治療法の開発に取り組んでいます。この治療法では、直線状の一本鎖DNAゲノムを持つ、ディペンドウイルス属の無害なアデノ随伴ウイルス(AAV)を使用し、シュワン神経細胞の機能に必要な神経栄養因子3(NT-3)タンパク質をコードするNTF3遺伝子を体内に導入します。
ヘリクスミスは、2020年末までに、CMTタイプ1の筋肉症状を治療するための韓国開発の遺伝子治療薬Engensis(VM202)の臨床試験を開始する予定である。[ 10 ]
防止
CMTの予防には、特に夫婦のどちらかにCMTの家族歴がある場合、将来の親への遺伝カウンセリングが効果的です。しかしながら、家族歴にCMTの病歴がないにもかかわらず、新生の点遺伝子変異が認められるケースも確認されています。
妊娠中には、絨毛生検(妊娠10~13週)と羊水検査(15~18週)によって、将来生まれてくる子供がシャルコー・マリー・トゥース病を発症する確率を調べることができます。
予測
一般的に、シャルコー・マリー・トゥース病の様々な病型の予後は臨床的重症度によって異なりますが、いずれの場合も病気はゆっくりと進行します。多くの患者は障害を抱えますが、それによって余命が縮まることはありません。