記事の医療専門家

新しい出版物
脳梁の形成不全
最後に見直したもの: 12.07.2025

すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
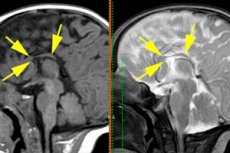
脳の両半球をつなぐ神経線維の接着が完全にまたはほぼ完全に欠如する発達障害は、脳梁形成不全症と定義され、これはその無形成症、つまり発達過程における形成の欠如と同義である。[ 1 ]
疫学
脳の先天性奇形は、個体発生の胚期におけるすべての異常の少なくとも 25% を占めます。
あるデータによると、適応症に基づいて脳の MRI 検査を受けた患者の 0.3 ~ 0.7 % に、脳梁無形成症 (形成不全) を含む脳梁体の異常が検出されます。
小児の脳梁体孤立性無形成症はまれな先天性欠損症ですが、遺伝的に決定される症候群の一部として、かなり一般的な先天異常であると考えられており、その有病率は発達障害のある小児 1 万人あたり 230 件と推定されています。
脳梁形成不全症または部分的無形成症の症例の 3 分の 1 では、精神障害が観察されます。
原因 脳梁の形成不全
脳梁体形成不全症は、脳半球間の情報伝達と協調機能を担う先天性欠損症であり、多くの場合、医師は正確な原因を特定できません。しかし、最もよくある原因は、胎児の脳構造の子宮内形成に影響を与える染色体異常、または脳形成異常を伴う遺伝性症候群の一部である遺伝性異常です。[ 2 ]
そのため、ワルカニー症候群、パトー症候群、エドワーズ症候群などの染色体過剰症候群(トリソミー)の場合には、胎児に脳梁が形成されません。
脳梁体欠損は、遺伝学的に確定診断されるモワット・ウィルソン症候群、アイカルディ症候群、マーデン・ウォーカー症候群、ドン・バロー症候群、アンダーマン症候群、プラウド症候群、アペール症候群、X連鎖性水頭症症候群において認められます。また、脳梁体の部分的形成不全は、ピット・ホプキンス症候群、ダンディ・ウォーカー症候群、センセンブレンナー症候群の特徴です。
脳梁の形成は、裂脳症などの脳回旋の異常、または先天性脳瘤や脳構造の嚢胞(チャドリー・マッカロー症候群の場合など)、ならびに奇形またはアーノルド・キアリ症候群の場合に阻害される。[ 3 ]
危険因子
脳梁形成不全症やその他の先天性脳欠損症の考えられる危険因子としては、放射線量の増加やさまざまな毒素による胎児への催奇形性作用、妊娠中のアルコールや薬物の摂取、特定の医薬品の使用、母親のウイルス感染などが挙げられます。
発達障害や脳形成不全の家族歴がある場合、子供にこの欠陥が生じるリスクも高まります。
病因
脳梁は妊娠6週から8週にかけて形成され始めますが、妊娠3週から15週の間にこの過程に障害が生じることがあります。発生学では、脳梁欠損の病因は2つの生物学的メカニズムに関連しています。
まず、背外側移動(神経堤(神経管の縁にある外胚葉の細胞帯)または頭部の中内胚葉から脳構造の形成部位への胚細胞の移動)を制御・調整する遺伝子の欠陥によって説明できる可能性があります。ほとんどの胚奇形や先天異常は、このプロセスの破綻によって生じます。
脳梁無形成症のもう一つのメカニズムは、大脳新皮質ニューロンの軸索が胎児の脳の両半球の中心線を横切らず、右半球と左半球の間に線維束を形成する代わりに、脳の両半球を繋ぐことなく縦方向に位置する異常な神経線維の束が形成されることであると考えられる。[ 4 ]
症状 脳梁の形成不全
合併症とその結果
脳梁体形成不全症は、関連する脳の異常に応じて、後遺症や合併症が異なります。最も重度の脳奇形を持つ小児では、てんかん発作、痙縮、水頭症、身体的および精神的発達障害が現れることがあります。
差動診断
脳梁体の他の病変(形成不全、部分的な発育不全、萎縮または低形成)を識別および鑑別するとともに、遺伝性症候群の存在を確認するために鑑別診断が行われます。[ 9 ]
連絡先
処理 脳梁の形成不全
脳梁を正常な状態に戻す方法はありません。そのため、治療としては、この欠陥による症状の重症度を軽減することが考えられます。
- 抗てんかん薬の使用;
- 理学療法、電気けいれん療法(筋力の増強と運動協調の改善)[ 10 ]
- 言語療法;
- 作業療法による基礎スキルの開発。
防止
予防策としては、さまざまな要因による催奇形性の影響の予防と先天性疾患の出生前診断のみが考えられます。
予測
一般的に、予後は脳梁体部形成不全の症状の程度と重症度、および付随する発達障害の有無によって決まります。[ 11 ]
この先天異常の軽度症例を持つ小児では、神経精神医学的悪影響は最小限で、ほぼ正常な機能を維持できる場合があります。また、成人になると、脳梁のない人でも平均的な知能を持ち、通常の生活を送る人もいます。