
小児の遺伝性腎炎(アルポート症候群
最後に見直したもの: 05.07.2025

すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
遺伝性腎炎(アルポート症候群)は、遺伝的に決定される遺伝性の非免疫性糸球体症であり、血尿(場合によってはタンパク尿を伴う)、慢性腎不全の発症を伴う腎機能の進行性低下として現れ、多くの場合、感音性難聴や視覚障害を伴います。
この疾患は1902年にLG・ガスリーによって初めて報告されました。ガスリーは、数世代にわたって血尿が認められる家系を観察しました。1915年には、AF・ハーストが同じ家系のメンバーに尿毒症が発症したことを報告しました。1927年には、A・アルポートが血尿を呈する複数の親族に難聴が見られることを初めて確認しました。1950年代には、同様の疾患による眼病変が報告されました。1972年には、遺伝性血尿患者の腎組織の形態学的研究において、ヒングレイスらが糸球体基底膜の不均一な拡張と重層化を明らかにしました。1985年には、遺伝性腎炎の遺伝学的根拠がIV型コラーゲン遺伝子の変異であることが特定されました(Fiengoldら、1985年)。
疾患の遺伝学的性質に関する研究により、遺伝性腎炎の表現型(難聴の有無を問わず)の違いは、変異遺伝子の発現レベルに起因するという結論に至りました。そのため、現在ではすべての臨床的変異は単一の疾患の症状とみなされており、「遺伝性腎炎」という用語は「アルポート症候群」と同義です。
疫学的研究によれば、遺伝性腎炎は10万人あたり17人の割合で発症します。
 [
[ アルポート症候群の原因
この疾患の遺伝学的根拠は、IV型コラーゲンのα5鎖遺伝子の変異です。この型は腎臓、蝸牛器、水晶体嚢、網膜、眼の角膜の基底膜に普遍的に存在し、このコラーゲン分画に対するモノクローナル抗体を用いた研究で証明されています。最近、遺伝性腎炎の出生前診断にDNAプローブを使用できる可能性が示唆されています。
変異遺伝子の保因者を特定するために、家族全員をDNAプローブで検査することの重要性が強調されており、これはこの疾患の家族に対する医療および遺伝カウンセリングにおいて非常に重要です。しかし、最大20%の家族には腎臓病を患う親族がおらず、これは異常遺伝子の自然変異の頻度が高いことを示唆しています。遺伝性腎炎の患者の多くは、家族内に腎臓病、難聴、視覚障害を持つ人がいます。血縁者同士の結婚では、両親から同じ遺伝子を受け継ぐ確率が高くなるため、1人以上の祖先を持つ人々との血縁結婚は重要です。常染色体優性、常染色体劣性、および優性、X連鎖性の遺伝経路が確立されています。
小児の場合、遺伝性腎炎は、アルポート症候群、難聴を伴わない遺伝性腎炎、家族性良性血尿の 3 種類に分けられます。
アルポート症候群は、難聴を伴う遺伝性腎炎です。腎臓、耳、眼の構造における糸球体基底膜のコラーゲン構造の複合的な欠陥が原因です。古典型アルポート症候群の遺伝子は、X染色体長腕の21-22q座に位置します。多くの場合、X染色体に連鎖する優性遺伝で受け継がれます。この点において、アルポート症候群は男性でより重症化します。これは、女性では変異遺伝子の機能が、損傷を受けていない2番目の染色体の健康な対立遺伝子によって補われるためです。
遺伝性腎炎の発症の遺伝学的根拠は、IV型コラーゲンα鎖遺伝子の変異です。IV型コラーゲンGには6つのα鎖が知られています。α5鎖およびα6鎖(Col4A5およびCol4A5)の遺伝子はX染色体長腕の21-22q領域に位置し、α3鎖およびα4鎖(Col4A3およびCol4A4)の遺伝子は第2染色体上、α1鎖およびα2鎖(Col4A1およびCol4A2)の遺伝子は第13染色体上に位置しています。
ほとんどの場合(80~85%)、X連鎖遺伝様式で本疾患が検出され、欠失、点突然変異、またはスプライシング障害によるCol4A5遺伝子の損傷が関連しています。現在、Col4A5遺伝子には200以上の変異が見つかっており、IV型コラーゲンのα5鎖の合成を阻害する原因となっています。この遺伝様式では、本疾患は男女ともに発症しますが、男児ではより重症化します。
IV型コラーゲンのα3鎖およびα4鎖の合成を担うCol4A3遺伝子およびCol4A4遺伝子座位の変異は、常染色体遺伝性です。研究によると、遺伝性腎炎の症例の16%に常染色体優性遺伝が認められ、6%に常染色体劣性遺伝が認められます。Col4A3遺伝子およびCol4A4遺伝子の変異には、約10種類のバリアントが知られています。
変異の結果、IV型コラーゲンの組み立てプロセスが阻害され、構造が破壊されます。IV型コラーゲンは、糸球体基底膜、蝸牛器、眼の水晶体の主要構成要素の一つであり、その病理は遺伝性腎炎の臨床において検出されます。
糸球体基底膜を構成するコラーゲンIV型は、主に2本のα1鎖(IV)と1本のα2鎖(IV)から構成され、さらにα3、α4、α5鎖も含んでいます。X連鎖遺伝において最もよく見られるのは、Col4A5遺伝子の変異により、コラーゲンIV型構造中のα3、α4、α5、α6鎖が欠損し、糸球体基底膜中のα1鎖とα2鎖の数が増加することです。この現象のメカニズムは不明ですが、mRNAの転写後変化が原因と考えられています。
アルポート症候群の初期段階では、糸球体基底膜のIV型コラーゲン構造におけるα3、α4、α5鎖の欠損により、IV型コラーゲンが菲薄化し脆弱化します。臨床的には、血尿(タンパク尿を伴う血尿やタンパク尿のみの症状は比較的少ない)、難聴、円錐水晶体が症状として現れます。病気がさらに進行すると、後期には基底膜の肥厚と透過性の低下が起こり、V型およびVI型コラーゲンの増殖が促進されます。その結果、タンパク尿の増加と腎機能の低下が認められます。
遺伝性腎炎の原因となる変異の性質は、その表現型の発現を大きく左右します。X染色体の欠失と、IV型コラーゲンのα5鎖およびα6鎖の合成を担うCol4A5遺伝子およびCol4A6遺伝子の同時変異が併発した場合、アルポート症候群は食道および性器の平滑筋腫症を併発します。研究データによると、欠失を伴うCol4A5遺伝子の変異の場合、この遺伝子の点変異と比較して、病理学的過程がより重篤であり、腎障害と腎外症状の併発、そして慢性腎不全の早期発症が認められます。
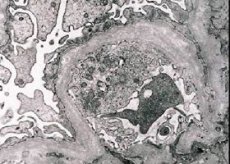
形態学的には、電子顕微鏡検査により、糸球体基底膜(特に緻密層)の菲薄化と重層化、および高電子密度顆粒の存在が明らかになります。糸球体病変は、微小な局所性メサンギウム病変から糸球体硬化症まで、同一患者において不均一に発現することがあります。アルポート症候群における糸球体炎は常に免疫陰性であり、この点で糸球体腎炎と鑑別されます。特徴的な所見としては、尿細管萎縮の発現、リンパ組織球浸潤、そして脂質封入体(リポファージ)を含む「泡沫細胞」の存在などが挙げられます。病気が進行するにつれて、糸球体基底膜の肥厚と顕著な破壊が明らかになります。
免疫系における特定の変化が明らかになりました。遺伝性腎炎の患者では、血中IgA濃度が低下し、IgM濃度が上昇する傾向があります。IgG濃度は病気の初期段階で上昇し、後期には低下することがあります。おそらく、IgMおよびG濃度の上昇は、IgA欠乏に対する一種の代償反応であると考えられます。
Tリンパ球系の機能活性が低下し、IgAの合成を担うBリンパ球の選択的減少が認められ、好中球における走化性と細胞内消化プロセスの破壊により、免疫の貪食結合が破壊される。
アルポート症候群患者の腎生検では、電子顕微鏡検査により糸球体基底膜の超微細構造変化が明らかになります。糸球体基底膜の菲薄化、構造の破壊、そして厚みの変化と不均一な輪郭を伴う分裂が見られます。遺伝性腎炎の初期段階では、この欠陥が糸球体基底膜の菲薄化と脆弱性の原因となります。
糸球体膜の菲薄化はより好ましい徴候であり、女児に多く見られます。遺伝性腎炎においてより顕著な電子顕微鏡的徴候は基底膜の裂傷であり、その破壊の程度は病状の重症度と相関します。
小児におけるアルポート症候群の症状
アルポート症候群の初期症状は、孤立性尿路症候群として、生後3歳までの乳幼児に最も多く見られます。ほとんどの場合、この疾患は偶然発見されます。尿路症候群は、乳幼児の予防検診、保育施設への入所前、または急性腎不全(ARVI)中に発見されます。ARVI中に尿に病変が認められた場合も同様です。遺伝性腎炎は、後天性糸球体腎炎とは異なり、潜伏期がありません。
病気の初期段階では、子供の健康状態はほとんど悪化しませんが、特徴的な特徴は尿路症候群の持続性と抵抗性です。主な兆候の1つは、重症度の異なる血尿であり、100%の症例で観察されます。血尿の程度の増加は、呼吸器感染症、身体活動、または予防接種後、またはその直後に認められます。ほとんどの場合、タンパク尿は1g /日を超えませんが、病気の初期には不安定になることがあります。病状が進むにつれて、タンパク尿は増加します。定期的に、リンパ球優位の白血球尿が尿沈渣に存在することがありますが、これは間質性変化の発生に関連しています。
その後、部分的な腎機能障害が発生し、患者の全身状態が悪化します。中毒、筋力低下、動脈性低血圧、多くの場合聴覚障害(特に男児)、そして時には視覚障害が現れます。中毒は、顔色の蒼白、疲労、頭痛として現れます。病気の初期段階では、ほとんどの場合、難聴は聴力検査でのみ検出されます。アルポート症候群の難聴は、小児期のさまざまな時期に発生する可能性がありますが、ほとんどの場合、難聴は6〜10歳で診断されます。小児の難聴は高周波から始まり、空気伝導と骨伝導でかなりの程度に達し、音伝導難聴から音知覚難聴に移行します。難聴は、この病気の最初の症状の1つである可能性があり、尿路症候群に先行する可能性があります。
アルポート症候群の患者の 20 % に、視覚器官の変化が見られます。最も頻繁に検出される異常は水晶体の異常で、球状水晶体、前部円錐水晶体、後部円錐水晶体または混合円錐水晶体、およびさまざまな白内障があります。アルポート症候群の家族では、近視の頻度がかなり高くなります。多くの研究者が、これらの家族で黄体の明るい白っぽいまたは黄色の顆粒の形で両側の黄斑周囲の変化を常に指摘しています。彼らはこの兆候を、アルポート症候群の高い診断価値を持つ一定の症状だと考えています。KS Chugh ら (1993) の眼科研究では、アルポート症候群の患者の 66.7% に視力低下、37.8% に前部円錐水晶体、22.2% に網膜斑、20% に白内障、6.7% に円錐角膜が認められました。
遺伝性腎炎の小児患者の中には、特に腎不全を発症すると、身体発達の著しい遅れが認められる場合があります。腎不全が進行すると、動脈性高血圧症を発症します。小児では、思春期以降や高齢期に発見されることが多いです。
遺伝性腎炎患者は、様々な(5~7種類以上)結合組織形成異常の徴候を呈することを特徴とします。患者にみられる結合組織形成異常の徴候の中で最も一般的なものは、眼間開離、高口蓋、咬合異常、耳介の異常な形状、手の小指の湾曲、そして足の「サンダルギャップ」です。遺伝性腎炎は、家系内での形態形成異常の徴候の均一性、および疾患が伝播した発端者の親族間での高頻度の分布を特徴とします。
疾患の初期段階では、アミノ酸輸送、電解質、濃縮機能、酸生成などの部分的な腎機能の低下が認められます。その後の変化は、ネフロンの近位部と遠位部の両方の機能状態に影響を及ぼし、複合的な部分的障害を特徴とします。糸球体濾過の低下は、より遅く、思春期に多く見られます。遺伝性腎炎が進行するにつれて、貧血が発生します。
このように、遺伝性腎炎は段階的な経過を特徴とします。まず、排尿症候群のわずかな変化として現れる潜伏期、あるいは隠れた臨床症状が現れ、その後、徐々に代償不全となり、腎機能の低下とともに顕在的な臨床症状(中毒、無力症、発達遅延、貧血)が現れます。臨床症状は通常、炎症反応の重層性に関係なく現れます。
遺伝性腎炎は、ある時期まで抑制された状態にある遺伝子の働きに依存し、さまざまな年齢で発症する可能性があります。
分類
遺伝性腎炎には3つのタイプがあります
- オプションI - 臨床的には、血尿、難聴、眼障害を伴う腎炎として現れます。腎炎の経過は進行性で、慢性腎不全へと進行します。遺伝形式はX染色体に関連する優性遺伝です。形態学的には、基底膜の構造異常、菲薄化、分裂が認められます。
- オプションII - 臨床的には、難聴を伴わない血尿を伴う腎炎として現れます。腎炎の経過は進行性で、慢性腎不全へと進行します。遺伝形式はX染色体に関連する優性遺伝です。形態学的には、糸球体毛細血管基底膜(特にラミナデンサ)の菲薄化が認められます。
- オプションIII:良性家族性血尿。経過は良好で、慢性腎不全は発症しません。遺伝形式は常染色体優性または常染色体劣性です。常染色体劣性遺伝の場合、女性ではより重篤な病状が認められます。
アルポート症候群の診断
以下の基準が提案されています。
- 各家族に少なくとも2人の腎症患者が存在すること。
- 発端者における腎症の主症状としての血尿
- 家族の少なくとも1人に難聴がある。
- 1 人以上の親族に慢性腎不全が発症すること。
様々な遺伝性疾患および先天性疾患の診断においては、包括的な検査アプローチが大きな意味を持ち、とりわけ、子供の家系図を作成する際に得られるデータに細心の注意を払います。アルポート症候群の診断は、患者に4つの典型的な徴候のうち3つが認められた場合に有効とみなされます。すなわち、家族における血尿および慢性腎不全の存在、神経感覚性難聴の存在、患者における視覚障害、生検の電子顕微鏡的特徴における糸球体基底膜の裂傷の兆候、その厚さの変化、および不均一な輪郭の検出です。
患者の検査には、臨床的および遺伝学的研究方法、病歴の的を絞った研究、診断上重要な基準を考慮した患者の全般的な検査を含める必要があります。代償段階では、遺伝的負担の存在、低血圧、多発性胚発生異常、尿症候群の変化などの症候群に焦点を当てることによってのみ病理を検出できます。代償不全段階では、重度の中毒、無力症、身体発達の遅れ、貧血などの腎外症状が現れる可能性があり、腎機能が徐々に低下するとともに現れ、悪化します。ほとんどの患者では、腎機能の低下とともに、酸およびアミノ生成の低下が観察されます。患者の50%は腎臓の分泌機能の大幅な低下に気づきます。尿の光学密度の変動範囲が制限されます。濾過リズムの乱れ、そして糸球体濾過の低下が見られます。慢性腎不全の段階は、患者の血清中の尿素濃度が 3 ~ 6 か月以上上昇し (0.35 g/l 以上)、糸球体濾過が正常値の 25% に低下した場合に診断されます。
遺伝性腎炎の鑑別診断は、まず血尿を伴う後天性糸球体腎炎から行うべきです。後天性糸球体腎炎は、感染後2~3週間で急性発症することが多く、発症初日から高血圧(遺伝性腎炎では低血圧)などの腎外症状がみられます。また、発症時には糸球体濾過量が低下し、部分尿細管機能障害はみられませんが、遺伝性腎炎ではこれらの症状がみられます。後天性糸球体腎炎は、より顕著な血尿とタンパク尿を呈し、赤沈値が上昇します。遺伝性腎炎の特徴である糸球体基底膜の典型的な変化は、診断上の価値があります。
代謝異常性腎症との鑑別診断は、慢性腎不全と家族内で臨床的に多様な腎疾患が認められる場合に行われます。腎盂腎炎から尿路結石症まで、腎症の症状は多岐にわたります。小児では、腹部の痛みや排尿時の周期的な痛み、尿沈渣中のシュウ酸の出現を訴えることがよくあります。
遺伝性腎炎が疑われる場合は、診断を明確にするために患者を腎臓専門科に紹介する必要があります。
何を調べる必要がありますか?
どのように調べる?
どのようなテストが必要ですか?
連絡先
アルポート症候群の治療
レジメンには、激しい運動と新鮮な空気への曝露の制限が含まれます。食事は完全タンパク質、脂肪、炭水化物を十分に含み、腎機能を考慮した完全な食事です。慢性感染巣の検出と治療は非常に重要です。以下の薬剤が使用されます:ATP、コカルボキシラーゼ、ピリドキシン(1日最大50mg)、塩化カルニチン。治療は年に2~3回行います。血尿には、イラクサ、チョークベリージュース、ノコギリソウなどの生薬が処方されます。
国内外の文献において、プレドニゾロンによる治療や細胞増殖抑制剤の使用に関する報告があるが、その効果を判断することは困難である。
慢性腎不全の場合、血液透析と腎臓移植が行われます。
遺伝性腎炎に対する特異的な(効果的な病因的)治療法はありません。すべての治療は、腎機能の低下を予防し、遅らせることを目的としています。
食事は、腎臓の機能状態を考慮し、バランスの取れた高カロリー食にする必要があります。機能障害がない場合は、子供の食事には十分なタンパク質、脂肪、炭水化物を含める必要があります。腎機能障害の兆候がある場合は、タンパク質、炭水化物、カルシウム、リンの摂取量を制限し、慢性腎不全の発症を遅らせる必要があります。
身体活動は制限されるべきであり、子供はスポーツを避けることが推奨されます。
感染患者との接触は避け、急性呼吸器疾患の発症リスクを低減する必要があります。慢性感染巣の衛生管理は不可欠です。遺伝性腎炎の小児には予防接種は実施されておらず、疫学的適応がある場合のみ予防接種が可能です。
遺伝性腎炎に対するホルモン療法および免疫抑制療法は効果がありません。シクロスポリンAおよびACE阻害薬を複数年にわたって長期投与することで、ある程度の有効性(タンパク尿の減少および疾患進行の遅延)が示唆されています。
患者の治療では、代謝を改善する薬剤が使用されます。
- ピリドキシン - 2~3 mg/kg/日を3回に分けて4週間投与する。
- コカルボキシラーゼ - 50 mgを隔日で筋肉内に合計10~15回注射します。
- ATP - 1 日おきに 1 ml を筋肉内に 10 ~ 15 回注射します。
- ビタミンA - 1000 IU/年/日を1回、2週間摂取する。
- ビタミン E - 1 回につき 1 mg/kg/日を 2 週間投与します。
このタイプの治療は、患者の全身状態の改善、尿細管機能障害の軽減に役立ち、1 年に 3 回のコースで実施されます。
レバミゾールは免疫調節剤として使用できます。1 日 2 mg/kg を週 2 ~ 3 回投与し、投与間隔は 3 ~ 4 日です。
研究データによると、高圧酸素療法は血尿や腎機能障害の重症度に良い影響を与えます。
遺伝性腎炎の最も効果的な治療法は、適切な時期に腎移植を行うことです。この場合、移植腎において病気の再発は起こりませんが、ごくまれに(約5%)、糸球体基底膜抗原に関連する腎炎が移植腎で発生することがあります。
有望な方向性としては、出生前診断と遺伝子工学療法が挙げられます。動物実験では、IV型コラーゲンα鎖の合成を担う正常な遺伝子を腎組織に導入すると高い効率が得られ、その後、正常なコラーゲン構造の合成が観察されます。
Использованная литература