
亜急性壊死性レア脳筋症
最後に見直したもの: 04.07.2025

すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
 [
[ 原因 リア症候群
この疾患は、主にピルビン酸代謝の破綻と呼吸鎖における電子伝達系の障害に起因する、エネルギー産生を担う酵素の欠損を基盤としています。呼吸鎖のピルビン酸脱水素酵素複合体(α-E1サブユニット)、ピルビン酸カルボキシラーゼ、複合体1(NAD-コエンザイムQ-還元酵素)、および複合体4(シトクロム酸化酵素)の欠損が発症します。
呼吸鎖のピルビン酸カルボキシラーゼ複合体1(NAD-コエンザイムQ-還元酵素)および複合体4(シトクロム酸化酵素)の欠陥は常染色体劣性遺伝で、ピルビン酸脱水素酵素複合体(α-E1サブユニット)の欠陥はX連鎖劣性遺伝で遺伝することが確立されています。ATPaseの6番目のサブユニットに影響を及ぼすmtDNAの点突然変異の場合、ミトコンドリア遺伝が典型的です。最も頻繁に発生するのは、mtDNAの位置8993におけるチミンからグアニンまたはシトシンへの置換に関連するmiscens突然変異です。あまり一般的ではないのは、mtDNAの位置9176における突然変異です。T8993G突然変異がNARP症候群の主な欠陥であるという事実により、これら2つの疾患の家族が報告されています。小児では、MERRF 症候群で発生する、位置 8344 の mtDNA の変異も報告されています。
変異mtDNAがミトコンドリアの大部分に蓄積すると、リー症候群の重篤な病態を呈すると考えられています。この病態のミトコンドリア起源においては、変異mtDNAは全ミトコンドリアの90%に認められます。病態形成は、細胞におけるエネルギー産生の障害と乳酸アシドーシスの発症に関連しています。
症状 リア症候群
この疾患の最初の兆候は、幼少期(1~3歳)に現れます。しかし、生後2週間や6~7歳で発症する症例も知られています。最初は、非特異的な障害が現れます:精神運動発達の遅れ、食欲不振、嘔吐、体重減少。その後、神経症状が増加します:筋緊張低下または筋緊張亢進への移行を伴うジストニア、ミオクローヌスまたは強直間代発作、四肢の振戦、舞踏アテトーゼ、協調運動障害、腱反射の低下、無気力、眠気。脳神経変性は進行性です。錐体路および錐体外路機能不全の症状が増加し、嚥下機能が低下します。視覚器官の変化としては、眼瞼下垂、眼筋麻痺、視神経萎縮、そして頻度は低いものの網膜色素変性がよく見られます。ときには肥大型心筋症を発症し、頻呼吸発作が現れることもあります。
まれに、急性脳症として進行することもあります。より典型的なのは慢性または亜急性の経過で、発症から数年後に致命的な転帰に至るケースです。急速な経過(数週間)の場合は、呼吸中枢の麻痺により死に至ります。
診断 リア症候群
血液生化学検査では、血液および脳脊髄液中の乳酸およびピルビン酸の蓄積、ならびに血中アラニン含有量の増加により、乳酸アシドーシスが明らかになります。ケトン体濃度も上昇することがあります。尿中には、乳酸、フマル酸などの有機酸の排泄量の増加が検出されます。血中および組織中のカルニチン濃度は、しばしば低下します。
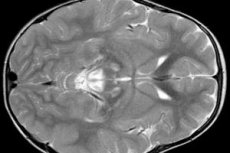
脳波検査の結果、てんかん活動の局所的な兆候が明らかになりました。MRI検査では、脳室の拡大、両側の脳損傷、基底核(尾状核、被殻、黒質、淡蒼球)の石灰化が明らかになりました。大脳半球および脳実質の萎縮も検出されます。
形態学的検査では、脳実質の著しい変化が明らかになります。主に脳中部、橋、基底核、視床、視神経において、対称性の壊死巣、脱髄、海綿状変性が認められます。組織学的所見としては、脳組織の嚢胞性変性、アストロサイトーシス、神経細胞死、細胞内ミトコンドリア数の増加が認められます。骨格筋では、脂質封入体の蓄積、呼吸鎖複合体1および4に対する組織化学反応の低下、筋線維膜下へのミトコンドリアの蓄積、クリステの崩壊を伴う異常なミトコンドリアが認められます。RRF現象はしばしば検出されません。
どのように調べる?
Использованная литература