
亜急性壊死性脳脊髄症
最後に見直したもの: 23.04.2024

すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
 [
[原因 レイア症候群
この疾患は、代謝障害、呼吸鎖におけるピルビン酸および欠陥電子輸送によって主に教育エネルギーを提供する、酵素欠損に基づくものです。ピルビン酸デヒドロゲナーゼ複合体欠損は、(-E1サブユニット)、ピルビン酸カルボキシラーゼ、錯体1(NADを補酵素Qレダクターゼ)と錯体4(シトクロムオキシダーゼ)呼吸鎖を開発しています。
これは、ピルビン酸デヒドロゲナーゼ複合体(-E1サブユニット)の欠陥は、欠陥ピルビン酸、錯体1(NADを補酵素Qレダクターゼ)と錯体4(シトクロムオキシダーゼ)、呼吸鎖が常染色体劣性様式で遺伝することがわかった - 劣性X連鎖。6-ATPアーゼサブユニットに影響を与えるときのmtDNA点突然変異、ミトコンドリア遺伝特性。ほとんどの場合、位置8993のmtDNAにグアニンまたはシトシンのチミンの交換に伴うmistsens変異を発生します。ミトコンドリアDNAの位置9176での稀突然変異。症候群NARPで基本的な欠陥は、これら2つの疾患の存在と家族に説明 - 。により変異T8993Gがあるという事実に 子どもたちはまた、シンドロームのMERRFに発見された位置8344、でのmtDNA変異を記載されています。
ミトコンドリアの大部分に突然変異mtDNAが蓄積した場合、重篤なLeia症候群が進行することが示唆されている。この状態のミトコンドリア発生において、突然変異mtDNAは全ミトコンドリアの90%で検出される。病原性は、細胞におけるエネルギー産生の侵害および乳酸アシドーシスの発症に関連する。
症状 レイア症候群
初期の年齢(1-3歳)での疾患デビューの最初の兆候。しかし、2週間で、年齢の6-7年で病気の症状の場合があります。精神運動遅滞、食欲不振、嘔吐エピソード、低体重:最初の非特異的障害を開発しました。その後の成長の神経症状では:筋肉の緊張低下やジストニア高張性への移行と、発作、ミオクローヌスまたは強直間代発作、手足の震え、舞踏、協調性障害、腱反射、無気力、眠気を減少させました。脳の神経変性は漸進的性質を有する。錐体と錐体外路疾患、嚥下障害の症状を拾い。多くの場合、眼瞼下垂、眼筋麻痺、視神経萎縮、網膜色素変性以下のように権威のような変更があります。時には、肥大型心筋症を開発し、頻呼吸のエピソードがあります。
まれに、この疾患は急性脳症のタイプに従って進行する。より特徴的なのは、慢性または亜急性の流れであり、この疾患の発症から数年後に致命的な結果に至る。急速な流れ(数週間)では、呼吸器の麻痺の結果として死が生じる。
診断 レイア症候群
生化学的血液検査では、乳酸アシドーシスは、血液中の乳酸およびピルビン酸の蓄積ならびに血液中のアラニン含有量の増加のために検出される。また、ケトン体のレベルを高めることができる。尿中では、有機酸の排泄が増加する:乳酸、フマル酸など。血液および組織中のカルニチン濃度はしばしば低下する。
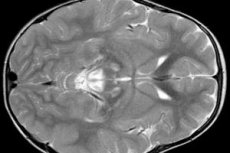
EEGの結果は、てんかん活動の焦点徴候を明らかにする。MRIデータによれば、脳室の拡張、両側脳損傷、基底核(尾状核、殻、黒色物質、淡蒼球)の石灰化が検出される。また、大脳半球および脳物質の萎縮を同定することも可能である。
脳の物質における形態学的研究ショー総変化:対称壊死、脱髄および脳の海綿状変性、主に中部、ブリッジ、大脳基底核、視床、視神経。組織学的画像には、脳組織の嚢胞性変性、星状細胞性神経膠症、ニューロンの死、細胞中のミトコンドリアの数の増加が含まれる。骨格筋において - 脂質封入体の蓄積は、ミトコンドリアsubsarkolemmalnoe輻輳の1、4、呼吸鎖、クリステの破壊と異常なミトコンドリアの複合体に組織化学的反応を減少させます。RRFの現象はしばしば検出されない。
どのように調べる?
Использованная литература