
角皮症:原因、症状、診断、治療
最後に見直したもの: 07.07.2025

すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
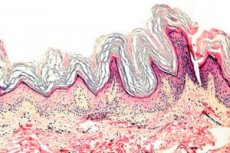
角化症は、角質化プロセスの障害(主に手のひらと足の裏の過剰な角質形成)を特徴とする皮膚疾患のグループです。
この疾患の原因と病態は完全には解明されていません。研究により、角化皮症はケラチン6、9、16をコードする遺伝子の変異によって引き起こされることが明らかになっています。ビタミンA欠乏症、特に性腺におけるホルモン機能不全、細菌感染およびウイルス感染は、病態において大きな役割を果たしています。これらは遺伝性疾患や内臓腫瘍(乾癬性角化皮症)の症状の一つです。
症状。びまん性角化症(ウンナ・トスト角化症、メレダ角化症、パピヨン・ルフェーブル角化症、切断性角化症、およびびまん性角化症を主症状の1つとして含む症候群)と局所性角化症(フィッシャー・ブッシュケの播種性斑状角化症、コスティの肢角膜弾性線維症、ブルハウアー・フランツェステスティの限局性角化症、フックスの線状角化症など)を区別します。
ヴィニー・トスト角化症(別名:先天性掌蹠・足底魚鱗癬、ヴィニー・トスト症候群)は、常染色体優性遺伝形式で遺伝します。生後2年以内に発症し、手のひらと足底(場合によっては足底のみ)の皮膚にびまん性の過剰な角質化が見られます。皮膚病変は、健康な皮膚との境界に青白い紅斑の帯として、手のひらと足底の皮膚がわずかに肥厚することから始まります。時間が経つにつれて、表面に滑らかで黄色がかった角質層が現れます。病変が手首や指の背に広がることは稀です。患者によっては、浅いまたは深い亀裂が生じ、局所的な多汗症が認められる場合があります。筆者が観察した患者では、母方の叔父、兄弟、息子がヴィニー・トスト角化症を患っていました。
ウィニー・トスト角化症における爪(肥厚)、歯、毛髪の損傷の症例が、さまざまな骨格異常および内臓、神経系、内分泌系の病変と組み合わさって説明されています。
組織病理学的検査:組織学的検査では、真皮上層に顕著な角化増殖、顆粒増生、表皮肥大、および小さな炎症性浸潤が認められる。鑑別診断:本疾患は他の種類の角化症との鑑別診断が必要である。
メレダ角化症(同義語:メレダ病、先天性進行性角化腫、シーメンス掌蹠角化症、コゴイ遺伝性掌蹠角化症)は、常染色体劣性遺伝性です。このタイプの角化症は、深い亀裂を伴う厚く黄褐色の角質層を特徴とします。病変の縁には、数ミリメートル幅の紫がかった境界線が見られます。この病変は通常、手足の甲、前腕、脛に広がります。多くの患者は局所性多汗症を経験します。この点では、手のひらと足の裏の表面がわずかに湿り、黒い点(汗腺管)で覆われます。
この病気は15~20歳で発症する可能性があります。爪は厚くなり、変形します。
組織病理学。組織学的検査では、角質増殖、時には表皮肥大、および真皮乳頭層の慢性炎症性浸潤が明らかになります。
鑑別診断。メレラ角化症はウンナ・トスト角化症と区別する必要があります。
パピヨン・ルフェーヴル角化症(同義語:歯周炎を伴う掌蹠角化症)は常染色体劣性遺伝します。
この病気は生後2~3年目に発症します。臨床像はメレラ病に類似しています。さらに、歯の変化が特徴的です(乳歯および永久歯の萌出異常、う蝕の発生、歯肉炎、急速に進行する歯周病、早期歯喪失)。
組織病理学。組織学的検査では、表皮の全層、特に角質層の肥厚と、真皮におけるリンパ球および組織球のわずかな細胞塊が明らかになりました。
鑑別診断:本疾患は他の角化症と鑑別する必要があります。重要な鑑別所見は、他の遺伝性びまん性角化症には見られない特徴的な歯の病理です。
角皮症(別名:フォンウィンケル症候群、遺伝性角化腫)は、常染色体優性遺伝性のびまん性角化症の一種です。生後2年目に発症し、多汗症を伴う手のひらと足の裏の皮膚にびまん性の角質沈着がみられます。時間の経過とともに、指に紐状の溝が形成され、拘縮や自然切断につながります。毛包性角化症は、手の甲、肘関節、膝関節周辺に現れます。爪甲は変化し(しばしば時計のガラス状になります)、性腺機能低下症、ルビー色脱毛症、難聴、爪厚肥厚などの症例が報告されています。
組織病理学。組織学的検査では、真皮に重度の角質増殖、顆粒腫、表皮肥大、リンパ球および組織球からなる小さな炎症性浸潤が認められます。
鑑別診断。切断性角化症を他のびまん性角化症と鑑別する際には、他のびまん性角化症では典型的ではない切断効果をまず考慮する必要があります。びまん性角化症のすべての形態の鑑別診断を行う際には、この症状が多くの遺伝性症候群の主な症状の一つとなる可能性があることを念頭に置く必要があります。
治療。ネオチガゾンは、角化症の一般的な治療に適応があります。投与量は症状の重症度に応じて、患者の体重1kgあたり0.3~1mgです。ネオチガゾンが使用できない場合は、ビタミンAを1日100~300,000mgの用量で長期間投与することが推奨されます。外用療法では、芳香族レチノイド、角質溶解剤、ステロイド剤を含む軟膏を使用します。
 [
[ あなたを悩ましているのは何ですか?
何を調べる必要がありますか?