
3型ムコ多糖症
最後に見直したもの: 04.07.2025

すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
 [
[ 病因
この疾患は、4つの異なる遺伝子の変異によって引き起こされます。リソソームα-N-アセチルグルコサミニダーゼ(ムコ多糖症III A)、アセチルCoA-α-グルコサミニド-N-アセチルトランスフェラーゼ(ムコ多糖症III B)、リソソームN-アセチルグルコサミン-6-スルファターゼ(ムコ多糖症III C)、およびスルファミダーゼ(ムコ多糖症III D)です。これらの酵素はすべてヘパラン硫酸の代謝に関与しています。
ヘパラン-N-スルファターゼ遺伝子(SGSH)は、17番染色体長腕(17q25.3)に位置しています。現在知られているSGSH遺伝子の変異の75.3%は点変異です。ヨーロッパ系集団に特徴的な頻度の高い変異として、R74C(ポーランドで56%、ドイツで21%)とR245H(オランダで56%)が報告されています。
R74C変異の頻度は47.5%、R245H変異は7.5%です。他の2つの変異、dell35GとN389Sは、合わせて変異アレルの21.7%を占めています。
α-N-アセチルグルコサミニダーゼ(NAGLU)遺伝子は、17番染色体長腕(17q21)に位置しています。NAGLU遺伝子に認められる変異の69%はミスセンス変異およびナンセンス変異であり、26.3%は小さな欠失および挿入です。アセチルCoA-α-グルコサミニド-N-アセチルトランスフェラーゼ(HGSNAT)遺伝子は、8番染色体短腕(8p11.1)に位置しています。この遺伝子は2006年に初めて特徴づけられ、現在までにわずかな変異しか見つかっていません。
N-アセチルグルコサミン-6-スルファターゼ遺伝子(GNS)は、12番染色体長腕(12ql4)に位置しています。世界ではムコ多糖症IIIDの患者が12名登録されています。GNS遺伝子には4つの変異が報告されています。
ムコ多糖症IIIのすべてのサブタイプにおいて、神経膜を含む細胞膜構造の一部であるヘパラン硫酸の分解障害が認められ、これは皮質萎縮によって引き起こされる重度の神経変性過程と相関しています。慢性下痢は、腸粘膜の機能不全とともに、病理過程における自律神経系の関与によって説明されます。感音難聴は、頻繁な中耳炎、耳小骨の変形、内耳の異常という3つの原因が考えられます。関節の硬直は骨幹端の変形の結果であり、関節包の肥厚はグリコサミノグリカンの沈着と線維化に続発します。症候群内における疾患の重症度の違いは、変異酵素の残存機能活性のみによるもので、活性が高いほど疾患は軽度です。
症状 3型ムコ多糖症
サンフィリッポ症候群における臨床的多型性は、他のムコ多糖症に比べてそれほど顕著ではありません。病気の進行は緩やかで、重度の神経障害と内臓やその他の器官系に現れる軽度の症状が特徴です。
この病気の最初の症状は、通常、それまで正常な発達をしていた小児に2歳から6歳の間に現れます。主な症状としては、精神運動発達および言語発達の退行、多動性症候群などの精神障害、自閉症または攻撃的な行動、睡眠障害、そして小児の不注意や不注意などが挙げられます。
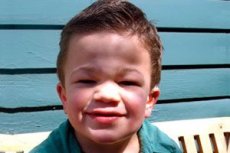
その他の一般的な症状は、多毛症、粗い髪、中等度の肝脾腫、四肢の外反変形、短い首です。ムコ多糖症III型では、ガーゴイル症などの粗い顔貌や、多発性骨異形成症などの骨格変形は、ハーラー表現型を特徴とする他のタイプのムコ多糖症と比較して、弱く発現します。身長は原則として年齢に比例し、関節の硬直が機能障害を引き起こすことはほとんどありません。多くの患者は骨粗鬆症や骨軟化症を発症することがよくあります。二次的な骨格障害は、病的骨折の危険性が高くなります。重度の精神神経疾患は、生後6~10年までに最も多く見られ、顕著な社会不適応につながります。進行性の感音難聴は、重症および中等度の疾患のすべての患者に固有のものです。病気が進行するにつれて、ほぼすべての患者にけいれんが観察されます。
この病気は急速に進行し、ほとんどの患者は20歳まで生存できません。ムコ多糖症IIIAは、この症候群の中で最も一般的で重篤なタイプであると考えられています。
診断 3型ムコ多糖症
ムコ多糖症IIIの診断は、尿中グリコサミノグリカン排泄量と酵素活性の測定によって確定されます。ムコ多糖症IIIでは、尿中グリコサミノグリカン総排泄量の増加とヘパラン硫酸の過剰排泄が認められます。ムコ多糖症IIIの特定のサブタイプに対応するリソソーム酵素の活性は、人工蛍光基質を用いて白血球または皮膚線維芽細胞培養物中で測定されます。
妊娠9~11週の絨毛生検における酵素活性測定、および/または妊娠20~22週の羊水中のグリコサミノグリカンスペクトルの測定により、出生前診断が可能です。遺伝子型が既知の家系では、妊娠初期にDNA診断を実施することも可能です。
どのようなテストが必要ですか?
差動診断
鑑別診断は、ムコ多糖症のグループ内と、ムコリピドーシス、ガラクトシアリドーシス、シアリドーシス、マンノシドーシス、フコシドーシス、GM1 ガングリオシドーシスなどの他のリソソーム蓄積疾患の両方で行われます。
処理 3型ムコ多糖症
現在まで、ムコ多糖症III型に対する効果的な治療法は開発されておらず、対症療法が適応となります。
Использованная литература