
脊髄性筋萎縮症
最後に見直したもの: 29.06.2025

すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
脊髄性筋萎縮症は、単一の病理学的単位ではなく、前脊髄角の運動ニューロンの変性過程の進行によって引き起こされる、臨床的および遺伝学的に異質な遺伝性病態の総体です。この用語は、脊髄運動ニューロンおよび/または脳幹の変性に起因する、遺伝的に規定された末梢麻痺および筋萎縮の様々な変異を包含します。最も一般的な原因は、第5染色体の長Q肩部位における常染色体劣性変異です。治療は非特異的であり、神経組織の栄養状態を改善し、生活の質を向上させるための緩和ケアを提供することを目的とします。[ 1 ]
疫学
脊髄性筋萎縮症は、新生児 6,000 ~ 10,000 人に 1 人の割合で発生します (American Journal of Medical Genetics 2002 によると)。
SMN 遺伝子エクソン 7 欠失保因者の有病率は 1:50 です。
球脊髄性筋萎縮症(ケネディ症候群)は、50,000 人に 1 人の小児に発生し、成人における最も一般的な脊髄性筋萎縮症のタイプです。
この病気に罹患した子供の半数は2年間の生存期間を越えられないことが指摘されています。
この病理は常染色体劣性遺伝の法則に従って遺伝します。多くの場合、病気の子の両親はそれぞれ変異遺伝子のコピーを1つずつ保有しています。この変異は2つ目の「正常な」遺伝子コピーの存在によって補償されるため、両親は脊髄性筋萎縮症の症状を呈しません。タイプ2の病理は通常、親から追加のコピーを受け継ぎません。この問題は、生殖細胞の形成過程、または受精直後の偶発的な障害によって発生します。タイプ1の脊髄性筋萎縮症では、自然発症は症例のわずか2%に過ぎません(この場合、両親のどちらか一方のみが保有者です)。[ 2 ]
原因 脊髄性筋萎縮症
脊髄性筋萎縮症の主な原因は、5番染色体qに位置するSMNタンパク質産生遺伝子の変異です。この疾患は、脊髄前角および脳幹の運動神経細胞の緩やかな死滅を引き起こします。これらの過程の結果として、筋緊張が低下し、呼吸筋、咽頭筋、顔面筋、骨格筋の萎縮が進行します。小児型脊髄性筋萎縮症の主な遺伝形式は常染色体劣性遺伝であり、両親が同時に欠陥遺伝子を保有することを意味します。IV型病態(成人型)はX染色体との関連があるため、男性のみが罹患します。
脊髄筋萎縮症の発症は、脊髄前角の運動ニューロンの変性と死滅、そして脳幹核の損傷の進行によって引き起こされます。病理学的変化は、頸部および腰椎部の肥厚部で最も顕著です。細胞数は最小限に減少し、細胞死プログラム(いわゆるアポトーシス)の失敗により結合組織への置換が起こります。この変化は、脳神経の運動核、前根、運動神経の構造に影響を及ぼします。神経原性束萎縮症の臨床像があります。病気の経過が長期化すると、末期には結合組織の過剰増殖が起こります。
対応する臨床像の出現は、脊髄前角の運動神経細胞の正常な機能に影響を与えるSMNタンパク質の欠乏と関連しています。脊髄性筋萎縮症の発症における一因としてタンパク質欠乏が発見されたのは、20世紀末のことです。運動ニューロンの損傷を背景に、骨格筋(主に近位部)の神経支配が障害されます。[ 3 ]
危険因子
脊髄性筋萎縮症 5q の臨床形態の多様性は、SMN タンパク質スコアに影響を及ぼす因子と、影響を及ぼさない因子の 2 つのカテゴリに分類できる特定の修飾因子の存在によって説明されます。
- 現在、SMN2遺伝子は脊髄性筋萎縮症の発症における基本因子と考えられており、SMN2遺伝子のコピー数が多いほど、疾患症状の強度は低下します。SMN遺伝子のセントロメアコピーに直接関連する2つ目の因子は、SMN2遺伝子のエクソン7における1ヌクレオチドの置換(c.859G>C)であり、この変異により新たなエンハンサー結合スプライスサイトが形成されます。その結果、SMN2遺伝子の転写産物にエクソン7が組み込まれます。この変異は、2型または3型の脊髄性筋萎縮症患者における全長SMNタンパク質の血中濃度の上昇と関連しています。
SMN の数に影響を与えるその他の要因:
- スプライシング制御因子 (Tra2β - エクソン 7 のエクソン スキッピングを誘導、SF2/ASF - エクソン 7 の包含を増加、hnRNPA1 - SMN2 遺伝子のエクソン 7 の包含を抑制)。
- 転写調節因子(CREB1 - SMN転写を増加、STAT3 - 軸索成長を促進、IRF1 - SMN数の増加、PRL - 重症段階での寿命の延長)。
- mRNA安定化因子(U1A-SMN、HuR/p38を減少させる)。
- 翻訳後修飾に影響を及ぼす因子 (RCA - SMN 分解を抑制、GSK3 - 生存率を向上)。
- 外因性要因(飢餓、低酸素症、酸化ストレス)。
上記の要因の影響は主にin vitroで判定されました。
- SMN 遺伝子に関連しない因子、特にシナプスでのエンドサイトーシスを最適化するタンパク質 (ラミニン 3、コロニン、ニューロカルシン デルタ、カルシウムニューリン様タンパク質)。
遺伝子発現の性質に影響を与える最も安定した修飾であるDNAメチル化にも注目が集まっています。病態に関与する可能性のある遺伝子群のメチル化は、脊髄性筋萎縮症の重症度と相関していることが判明しました。[ 4 ]
病因
脊髄性筋萎縮症は、常染色体優性遺伝、常染色体劣性遺伝、またはX連鎖遺伝のいずれかの遺伝形式を先天的に有する遺伝病態です。多くの場合、ここで言及されているのは幼児期に発症する常染色体劣性遺伝病態です。このような脊髄性筋萎縮症の形成に関与するのは、5q13遺伝子座に位置するSMN遺伝子です。SMN遺伝子のエクソン7の欠失は、近傍のp44遺伝子およびNAIP遺伝子の関与を示唆する病態を引き起こします。
SNMゲノムは、294個のアミノ酸から構成され、分子量約38 kDaのタンパク質をコードしています。このタンパク質は以下の機能を有します。
- RNA-タンパク質複合体の一部です。
- プレRNAスプライシングを触媒するスプライソソーム部位の形成に関与します。
- タンパク質の生成とタンパク質アイソフォームを制御するプロセスに関与します。
- mRNAの軸索輸送を提供します。
- 神経細胞の成長を促進し、神経筋伝達を促進します。
SMN 遺伝子にはいくつかの種類が知られています。
- テロメアSMNt(SMN1);
- セントロメアSMNc(SMN2)。
脊髄性筋萎縮症の症例のほとんどは、SMN1 遺伝子の変異が原因です。
ケネディ脊髄性筋萎縮症は、アンドロゲン受容体タンパク質をコードするNR3C3遺伝子を含むXq12遺伝子座に連鎖しています。X連鎖遺伝の変異を有し、遺伝子エクソン内のCAGリピート数が増加すると病態が進行します。
SNM タンパク質生成の抑制には、次のような変化が伴います。
- 軸索の協調が損なわれると、軸索の過剰な分岐が起こります。
- 軸索の成長が遅くなり、軸索のサイズも減少します。
- 成長円錐内のカルシウムチャネルが不適切にクラスター化している。
- 運動神経細胞の軸索の不規則な交感神経前終末が形成される。
脊髄は前角の運動ニューロンを活発に失い始め、それが四肢近位筋の萎縮の発症の原因となる。[ 5 ]
症状 脊髄性筋萎縮症
脊髄性筋萎縮症(SMA)の症状は、新生児期から生後6ヶ月までに現れることが多く、「無気力」な乳児症候群として現れます。胸郭の湾曲、強い筋緊張低下、反射消失、舌筋の痙攣、呼吸困難などが認められます。罹患した乳児は2歳未満で死亡することが多く、致死的な転帰は、感染過程の持続を背景に呼吸不全が悪化することによって引き起こされます。
脊髄性筋萎縮症の中間型である第2型は、生後6ヶ月から発症します。「無気力」な子どもの症状に加え、低血圧、反射神経の低下、呼吸障害、舌の痙攣がみられます。座ることができたとしても、大関節の複数の拘縮がみられます。
クーゲルベルク・ヴィーランダー脊髄性筋萎縮症も、幼児期に発症し、自力で動ける子どもの頃から発症します。腸骨筋、大腿四頭筋、内転筋の筋力低下、低血圧、反射神経の低下、舌の痙攣などがみられます。多くの患者は、年月とともに自力で動く(歩く)能力を失います。
脊髄性筋萎縮症4型は高齢で発症し、進行が緩やかで比較的予後が良好であることが特徴です。[ 6 ]
ケネディ萎縮は中年期に最も多く発症します(一般的に15~60歳)。症状としては、筋肉痛、筋力低下、女性化乳房、遠位筋の筋力低下、無気力、舌の痙攣、萎縮などが挙げられます。球麻痺の徴候も認められます。
- 嚥下困難;
- 願望;
- 咀嚼筋の衰弱;
- 構音障害;
- 手の姿勢および運動の震え。
アンドロゲン欠乏症の最初の兆候:
- 女性化乳房(患者の約 60%)、非対称であることが多い。
- 性機能の低下(精子減少症、精巣萎縮、勃起不全)。
最初の兆候
脊髄性筋萎縮症は、筋力低下と全身性インポテンスとして現れます。感覚機能や知的能力は影響を受けません。
神経筋病理学の主な指標:
- 筋肉が「怠惰」、弱くなり、筋肉の弛緩や緩みが認められます。
- 筋緊張は低下し、腱反射は最小限に抑えられるか、または消失します。
- 足底反射が正常または欠如している。
- 個々の筋肉群の短いけいれんが認められます(皮膚の下、舌の上に確認できます)。
- 筋萎縮の兆候が見られます。
ウェルドニッヒ・ホフマン症候群は、顕著な筋緊張低下、全身倦怠感、頭を支えられない、寝返りを打てない、起き上がることができないといった症状を呈します。赤ちゃんを腹部にぶら下げた状態で支えようとすると、体が「たわむ」ように見えます。咳、嚥下、吸啜反射は不十分で、食物が気道に入りやすく、呼吸に問題が生じます。子宮内筋緊張低下に伴い、関節の歪みが生じることもあります。妊娠中の既往歴では、胎児活動の低下が示唆されることが多いです。
脊髄性筋萎縮症I型の基本的な兆候:
- 運動発達の重度の遅れ。
- 関節拘縮および胸椎湾曲の急速な発症。
- 呼吸器系および眼球障害の増加、嚥下障害(食物および唾液の両方)および痰の排出障害。
- 誤嚥性炎症のリスク増加
- 感染症、進行性呼吸不全。
脊髄性筋萎縮症II型は、明らかな運動発達の阻害を呈します。多くの患者は介助なしで座ることができ、時には這ったり立ったりすることも可能ですが、これらの能力は時間の経過とともに失われることが多いです。指の震え、筋肉や関節(骨)の歪み、呼吸器系の問題が認められます。ふくらはぎの偽性肥大がみられる場合もあります。
タイプ II 病理の主な特徴:
- すでに獲得した技能や能力の発達の停止や逆行を含む発達の遅れ。
- 肋間筋の衰弱が進む;
- 横隔膜呼吸の浅さ、咳反射の弱まり、呼吸不全の徐々に悪化。
- 胸郭および脊柱の湾曲、拘縮。
クーゲルベルク・ヴィーランダー症候群では、症状はより軽度で、ゆっくりと進行します。患者は移動は可能ですが、ジョギングや階段の昇降には支障をきたします。遅発性の症状としては、嚥下困難や咀嚼困難などが挙げられます。
脊髄性筋萎縮症IV型は、高齢(成人)になってから発症し、最も「軽度」で良好な経過をたどることが特徴です。主な徴候は、運動能力の漸進的な喪失です。[ 7 ]
フォーム
脊髄性筋萎縮症は、変性変化、脊髄前角の運動神経細胞、そしてしばしば脳幹の運動神経核の死を特徴とする遺伝性疾患群の一つです。この病態は人生の様々な時期に発現することがあり、臨床像は必ずしも同じではありません。遺伝形式や病状の進行も異なります。
小児脊髄性筋萎縮症は、19世紀後半に初めて報告されました。20世紀半ば頃には、この疾患の主な病型が特定されました。
- 先天性(乳児の誕生直後に発症する)
- 早期乳児型(乳児のそれまでの正常な発達を背景に発生する)
- 後期乳児型(2歳以上で発症する)。
専門医の中には、2 番目と 3 番目の形態を 1 つの小児脊髄筋萎縮症としてまとめる人もいます。
病理は小児型と成人型に分けられることが一般的です。小児の脊髄性筋萎縮症は、早期型(生後数ヶ月以内に発症)、後期型、思春期型(青年期型または若年型)に分類されます。最もよくみられる症候群は以下のとおりです。
- ウェルドニッヒ・ホフマン萎縮症;
- クーゲルベルク-ヴィーランダー形式。
- 慢性乳児脊髄性筋萎縮症;
- ヴィアレット・ファン・ラーレ症候群(聴覚喪失を伴う延髄脊髄型)
- ファジオ・ロンデ症候群。
成人型脊髄性筋萎縮症は16歳以上から60歳頃までの間に発症し、比較的良好な臨床像と予後を呈します。成人の病態には以下のものがあります。
- ケネディ球脊髄萎縮症;
- 肩甲腓骨筋萎縮;
- 顔面-膝-肩型および眼咽頭型。
- 遠位脊髄萎縮;
- 単肢性脊髄萎縮症。
脊髄萎縮症は、単独型と複合型に分けられます。単独型は、脊髄運動ニューロンの障害が優位であることが特徴です(多くの場合、これが唯一の症状です)。複合型はまれであり、神経学的および身体的な障害が複合して現れます。先天性冠動脈奇形、聴覚機能障害、寡頭症、小脳低形成を伴う複合症候群の症例が報告されています。
高齢者の脊髄性筋萎縮症は、ケネディ球脊髄性筋萎縮症が最もよく代表されます。この病態はX連鎖性劣性遺伝です。病状の進行は緩やかで、比較的良性です。下肢近位筋の萎縮から始まります。手や頭の震えがみられる場合もあります。同時に、精巣萎縮、女性化乳房、糖尿病といった内分泌疾患も認められます。しかしながら、成人では小児よりも病状の進行は軽度です。
脊髄性筋萎縮症の異型。 |
病理学のデビュー |
検出可能な問題 |
死亡年齢 |
特徴的な症状 |
脊髄性筋萎縮症1型(別名:ヴェルディング・ホフマン脊髄性筋萎縮症) |
生後6ヶ月まで |
赤ちゃんは座ることができない |
最長2年間 |
重度の筋力低下、筋緊張低下、頭を支えるのが困難、泣きや咳ができない、嚥下や唾液分泌に問題があり、呼吸不全や誤嚥性肺炎を発症する |
脊髄性筋萎縮症2型 |
6ヶ月から1年半 |
赤ちゃんは立つことができない |
2年以上 |
運動遅滞、体重減少、咳の弱さ、手の震え、脊椎の湾曲、拘縮 |
脊髄性筋萎縮症3型(別名クーゲルベルク・ヴェランダー脊髄性筋萎縮症) |
1年半後。 |
最初は立ったり歩いたりできるが、ある年齢になるとこの能力が失われる可能性がある |
成人期。 |
筋力低下、拘縮、関節可動性亢進 |
脊髄性筋萎縮症4型。 |
思春期または成人期 |
最初は立ったり歩いたりできるが、ある年齢になるとこの能力が失われる可能性がある |
成人期。 |
近位筋の筋力低下、腱反射の低下、筋のけいれん(線維束性収縮)の増加 |
遠位脊髄萎縮とは、下半身を支配する脊髄の運動神経細胞の病変を指します。この病変の特徴的な兆候は以下のとおりです。
- 大腿筋の萎縮;
- 膝、足伸筋、股関節内転筋の筋力低下。
腱反射に変化なし。
遠位脊髄性筋萎縮症は、重複する表現型を持つ 2 つの対立遺伝子変異によって表されます。
- 肩甲会陰脊髄性筋萎縮症;
- 遺伝性シャルコー・マリー・トゥース病 2C 型運動感覚ニューロパチー。
近位脊髄性筋萎縮症5qは、弛緩性麻痺と筋萎縮の症状が進行するのを特徴とし、これは脊髄前角のα運動ニューロンの変性変化によるものです。産後仮死を伴う先天性疾患は最も重篤な病態であり、出生直後から運動活動は実質的に停止し、拘縮、嚥下障害、呼吸障害がみられます。このような乳児はほとんどの場合、死亡に至ります。
合併症とその結果
脊髄性筋萎縮症がさらに進行すると、四肢(特に脚)の筋力低下と筋量減少が起こります。乳児は当初は獲得した能力を全く発揮できず、あるいは徐々に失っていきます。つまり、歩行能力や支えなしで座る能力などを失います。上肢の運動機能が低下し、関節が硬くなり、時間の経過とともに拘縮が生じ、脊柱が湾曲していきます。
運動能力をできるだけ長く維持し、合併症の発症を防ぐために、次のことが推奨されます。
- ベッドにいるとき、座っているとき、歩いているときなど、正しい姿勢(反重力姿勢)を練習します。
- 脊髄性筋萎縮症の種類に関係なく、定期的な理学療法、ストレッチ運動、マッサージ、理学療法。
- 特別なベッド、椅子(車椅子)、マットレス、枕を使用します。
- サポート用の矯正器具やコルセットを選択して使用します。
- 呼吸器系、筋骨格系、消化器系、神経系、心臓血管系に良い効果をもたらすハイドロセラピーとキネシオセラピーを実践します。
- 臨床検査、脊椎および骨盤のレントゲン撮影を含む定期的な診断検査を実施します。
- 同様の患者を扱った経験のある理学療法士や整形外科医に体系的に相談してください。
- 状況に応じてコルセット、装具、整形外科用器具、車椅子などを調整します。
脊髄性筋萎縮症患者の介護者は以下の点について理解しておく必要があります。
- 安全な行動、理学療法、マッサージ、物理療法の基礎を備え、
- 患者の独立した活動を維持する規則に従って、整形外科用装置を使用します。
- ケア、衛生のルールを守ります。
脊髄性筋萎縮症は、咀嚼、嚥下、食物の輸送に障害を伴うことが多く、誤嚥の危険性が高まり、誤嚥性肺炎や気道閉塞といった、第一型病態の最も特徴的な症状が出現します。嚥下障害は、食事時間の著しい持続的な延長、食事への抵抗、口からの食物の落下、頻繁な嘔吐、体重減少の悪化といった症状によって示されます。
消化管運動障害は、便秘、蠕動運動の弱化、胃内での食物の長時間滞留(胃停滞)、胃食道逆流症の発症といった形で現れます。こうした合併症を予防するためには、以下の対策が必要です。
- 食事中の患者の正しい姿勢を監視します。
- 必要に応じて、胃チューブまたは胃瘻を使用して十分な水分と栄養摂取を確保し、誤嚥のリスクを減らします。
- 食べ物や飲み物の準備に関するルールを守り、その一貫性や食事の頻度に注意してください。
- 医師の処方に応じて、薬物療法、マッサージ、理学療法などを行います。
脊髄性筋萎縮症の最も深刻な合併症の一つは、呼吸筋の筋力低下に伴う呼吸器系機能障害です。呼吸器疾患は、1型病態の乳児、2型または3型病態の青年期および成人のいずれにおいても致命的となる可能性があります。主な問題点は以下のとおりです。
- 咳反射が障害され、呼吸器からの痰の排出に問題が生じます。
- 肺に入る空気量の減少が進み、肺からの二酸化炭素の排出が阻害される。
- 胸部が歪み、肺が圧迫され変形します。
- 気管支肺炎の形で起こる感染過程。
このような合併症を予防するために、患者にはアンビューバッグを使った呼吸訓練を行うことが推奨されることが多い。[ 9 ]
診断 脊髄性筋萎縮症
脊髄筋萎縮症が疑われる患者の場合、次のような検査が診断上の価値があります。
- 血液化学;
- 遺伝子DNA分析;
- 電気筋電図検査。
追加の方法としては、筋線維の生検、筋肉と脳の超音波および共鳴画像検査を実施することも可能です。
血液検査ではクレアチンホスホキナーゼが生理的に正常と示されることもありますが、場合によっては約 2.5 倍に上昇することもあります。
電気筋電図は、脊髄運動ニューロンの喪失による変化を明らかにする。これは、干渉曲線の振幅の減少、特定の「周波数リズム」を形成する線維性収縮および線維束性収縮である自発活動電位の発生によって検出される。末梢運動線維を通過するインパルス信号の速度は正常であるか、二次的な脱神経障害により低下する。[ 10 ]
機器診断では、筋肉の超音波検査やMRI検査もしばしば用いられ、脂肪組織による筋肉の置換を検出できます。MRI検査では、脊髄性筋萎縮症特有の典型的な病理学的過程パターンが明らかになりますが、これは病変の後期にのみ可能です。
患者の筋生検における形態学的解析では、筋束萎縮と筋線維の集塊という非特異的な像が認められる。罹患筋線維の大部分はタイプ1に属し、免疫組織学的および化学的特徴は正常範囲内である。超微細構造像は非特異的である。
脊髄性筋萎縮症の疑いがある場合、最も重要な診断手順は、SMN遺伝子変異を検出できる検査です。直接DNA分析により、SMNc遺伝子とSMNt遺伝子の第7エクソンと第8エクソンの有無を検出できます。最も有益な方法は定量分析であり、遺伝子コピー数を決定することで脊髄性筋萎縮症の病態を解明することができます。定量分析は患者の状態を評価する上でも重要であり、さらなる医学的および遺伝学的家族カウンセリングのために実施される必要な手段です。
SMN遺伝子欠失の陰性結果が得られた後にのみ、追加の診断検査を実施します。点変異の検出が必要な場合は、SMNt遺伝子の直接自動シークエンシングが使用される場合があります。
差動診断
鑑別診断は、「無気力患者」の症状複合体を明らかにする病理学的過程、先天性筋ジストロフィー、構造性ミオパチーまたはミトコンドリア性ミオパチーの有無に基づいて行われます。特に、以下の病態の存在を除外する必要があります。
- 運動ニューロン疾患;
- 原発性側索筋硬化症;
- 筋ジストロフィー;
- 先天性ミオパチー;
- グリコーゲン蓄積に関連する疾患。
- ポリオ;
- 自己免疫性重症筋無力症。
診断アルゴリズムは、個々の小児の症状の特異性に応じて開発されます。そのため、機能状態に応じて特別な患者分類が用いられます(ユーロプロトコルTREAT-NMD)。
- 支えがなければ座ることができない(寝たきり)。
- 座ることはできるが歩くことはできない(座位)。
- 自力で移動できる(歩行可能な患者)。
最初のグループの患者には、次の診断アルゴリズムが推奨されます。
- 身体検査(胸郭の湾曲の検出、呼吸機能および咳嗽機能の評価、皮膚の状態)
- 心臓および呼吸のモニタリング、睡眠ポリグラフ検査、および肺換気不全の症状の特定。
- 酸素化の程度を決定するためのパルスオキシメトリー。
- 6 か月間の極端な期間中の感染性炎症性病変の頻度と抗生物質投与コースの評価。
- 胸部X線検査と反復動態検査。
- 嚥下機能の評価。
2 番目のグループの患者には、次のアルゴリズムが適用されます。
- 身体検査;
- 心臓と呼吸のモニタリング、肺換気不足を検出するための睡眠ポリグラフ検査。
- パルスオキシメトリー;
- 6 か月間の極端な期間中の感染性炎症プロセスと抗生物質投与の頻度の評価。
- 脊柱の検査、脊椎のX線検査、湾曲度の評価。
3 番目のグループの患者は、次のような研究の対象となります。
- 身体検査;
- 呼吸機能検査(スパイロメトリー、肺活量計算、呼吸筋機能の評価を含む)
- 極端な年間期間中の感染性炎症性病変および抗生物質投与の頻度を調べる。
SMN1遺伝子とSMN2遺伝子の類似性により、鑑別診断の実施が複雑になる場合があります。誤りを避けるため、SMN1遺伝子のエクソン7のコピー数を検出できるMLPA法の使用が推奨されます。
脊髄性筋萎縮症のほとんどの症例では、SMN1遺伝子のエクソン7および/または8のホモ接合性欠失が認められます。しかし、他の遺伝子(ATP7A、DCTN1、UBA1、BSCL2、EXOSC3、GARSなど)も原因となる可能性があり、SMN1検査が陰性の場合は注意が必要です。
研究に用いる生体材料は、末梢血、胎児血、乾燥血液スポットマップなどです。診断は必須です。
- 脊髄性筋萎縮症の重篤な病歴がある場合;
- 遺伝歴に関係なく、疑わしい症状が検出された場合。
さらに、妊娠を計画する責任があるすべてのカップルにも研究が推奨されます。
連絡先
処理 脊髄性筋萎縮症
脊髄性筋萎縮症の患者には、次のような包括的な治療が必要です。
- ケア、ヘルプ、サポート。
- ダイエット食品;
- 薬物療法;
- 運動療法や理学療法などの非薬物療法によるリハビリテーション措置。
筋骨格系だけでなく、身体のすべてのシステムに多様な効果をもたらす治療計画が標準です。
残念ながら、脊髄筋萎縮症を根本的に治癒することは不可能です。しかし、アミノ酸やマルチビタミン複合体、神経栄養薬、カルシウムチャネル遮断薬、血管拡張薬、強心薬および細胞増殖抑制薬、プロテアーゼ阻害剤、ステロイド薬、抗酸化剤、免疫グロブリンおよび免疫抑制剤などを適切に使用することで、患者の生活の質を向上させることは可能です。幹細胞、神経保護化合物、筋力強化分子を用いた治療は、予測不可能な全身性疾患を引き起こす可能性があることが実験的に証明されています。同時に、これらの治療適用後の良好な動態は、これまでのところ証明されていません。
この問題は正常なSMNタンパク質の欠乏によって引き起こされるため、SMNタンパク質レベルを25%以上増加させることで症状の改善が期待できます。そのため、ガバペンチン、リルゾール、ヒドロキシウレア、アルブテロール、バルプロ酸、フェニル酪酸ナトリウムなど、このタンパク質の産生を活性化する薬剤の研究が活発に行われています。
現代医学では、脊髄性筋萎縮症の外科的治療も行われています。これは、脊柱の外科的アライメント、つまり神経筋の弯曲矯正を目的としています。外科医は特殊な構造を用いて、脊柱を多段階に固定します。仙骨、骨盤、上部胸椎などの椎骨が支持点として用いられます。この手術は、脊柱のアライメントを整え、脊柱にかかる負荷を均等に分散させ、体位を変える際の不快感を解消し、内臓(肺を含む)への悪影響を回避するのに役立ちます。
医薬品
現在、脊髄性筋萎縮症の病因的治療法は確立されておらず、科学医学はこの課題に取り組んでいます。これまでに、科学者らはSMN2遺伝子からのmRNA産生を促進する薬剤の単離に成功しています。しかし、脊髄性筋萎縮症患者を対象とした大規模な国際臨床試験はまだ実施されていません。
標準的な治療計画に含まれる薬剤のほとんどは、一般的な作用原理を持ちますが、有効性の証拠は比較的低いです。
L-カルニチン |
天然アミノ酸で、ビタミンB群の「類似体」です。体内で生成され、肝臓と横紋筋に存在し、ビタミン類似物質の一種です。代謝プロセスに関与し、CoAの活性をサポートし、代謝を正常化するために用いられます。同化作用、抗甲状腺作用、抗低酸素作用を有し、脂質代謝と組織修復を促進し、食欲を最適化します。L-カルニチンは1日あたり約1,000mgを服用します。治療期間は最長2ヶ月です。 |
コエンザイムQ10(ユビキノン) |
複数のイソプレニル基を含むベンゾキノン基を持つ補酵素です。これらは脂溶性の補酵素で、主に真核生物の細胞構造のミトコンドリアに存在します。ユビキノンは電子伝達系に含まれ、酸化リン酸化に関与します。この物質はエネルギーを豊富に含む臓器、特に肝臓と心臓に最も多く存在します。コエンザイムQ10は抗酸化作用を有し、α-トコフェロールの抗酸化能を回復させます。通常、1日30~90mgを2ヶ月間服用します。 |
セレブロリジン |
神経栄養作用を有する向知性薬です。血管性認知症や脳卒中などの神経疾患の治療レジメンでよく使用されます。活性成分には、限界分子量1万ダルトンのペプチドが含まれています。本剤は1~2mlの静脈内注射で投与されます。治療コースは10~15回の注射で構成されます。 |
アクトベジン |
本剤は低分子量ペプチドとアミノ酸誘導体を主成分としています。アクトベジンは血液誘導体であり、限外濾過透析によって分離されます。本剤の使用により、酸素の吸収と利用が増加し、エネルギー代謝が促進されます。本剤は1~2mlの静脈内注射で投与され、10~15回の注射が必要です。 |
ソルコセリル |
脱タンパク化された血液透析液で、細胞前における酸素およびグルコース輸送の最適化、細胞内ATP産生の促進、組織再生反応の刺激、線維芽細胞の増殖の活性化、血管壁におけるコラーゲン産生の促進が期待されます。治療コースは、薬剤(1日1~2mL)を10~15回筋肉内注射することで構成されます。 |
ニューロマルチビット(ビタミンB複合体) |
マルチビタミンは、ビタミンB群の欠乏に積極的に用いられます。ビタミン剤の注射療法の代替として、優れた効果を発揮することがよくあります。脳内の代謝プロセスを活性化し、神経系組織の修復を促進し、鎮痛作用があります。ニューロマルチビタミンは、1日1~2錠を4週間または8週間服用してください。 |
ビタミンE |
よく知られている抗酸化作用のある脂溶性ビタミンです。1日10~20IUを1~2ヶ月かけて服用します。 |
バルプロ酸 |
鎮静作用およびリラックス作用を有し、抗けいれん作用を示し、中枢神経系におけるGABA濃度を高めます。1歳以上の小児の治療にのみ、1日10~20mg/kgを投与してください。 |
サルブタモール |
選択的β2アドレナリン受容体作動薬のグループに属する気管支拡張薬です。本剤の定期的な使用は、mRNAおよびSMNタンパク質の産生を増加させ、脊髄性筋萎縮症の臨床像に好ましい影響を与えます。サルブタモールは、1回2~4mgを1日4回(最大用量は1日32mg)慎重に投与します。 |
脊髄性筋萎縮症に使用される最新の薬剤の一つに、ゾルゲンスマ遺伝子治療薬があります。ゾルゲンスマは、導入された運動神経細胞の活性と正常な機能を確保します。この薬剤は、特別なプロトコルに従って免疫調節薬と併用され、1.1× 1014 Vg/kg(投与総量は患者の体重に応じて決定されます)の用量で、単回静脈内投与されます。
ゾルゲンスマによる治療を開始する前に、有効な診断法を用いてAAV9に対する抗体レベルを測定し、肝機能(ALT、AST、総ビリルビン)を評価し、一般臨床血液検査およびトロポニンI検査を実施し、クレアチニン値を測定することが必須です。急性および慢性の活動性感染症が検出された場合、薬剤の投与は感染症の治癒または再発期の完了まで延期されます。
この薬の最も頻繁な副作用は肝不全であると考えられており、これは致命的となる可能性があります。
脊髄筋萎縮症に対して医師が処方する可能性のあるその他の承認薬:
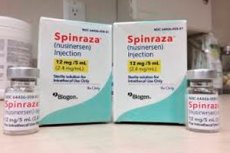
- スピンラザは、脊髄性筋萎縮症の治療に特化して設計されたアンチセンスオリゴヌクレオチドであるヌシネルセンナトリウムの製剤です。腰椎穿刺による脊髄内投与を目的としています。推奨用量は12mgです。治療レジメンは主治医が決定します。
- リスジプラムは、運動神経細胞生存遺伝子2のmRNA前駆体のスプライシングを修飾する薬剤です。リスジプラムは1日1回経口投与します。投与量は、患者の年齢と体重を考慮して医師が個別に決定します。生後2ヶ月未満の乳幼児への使用は禁忌です。この薬剤には胚・胎児毒性が認められているため、妊娠可能な患者は、投与中および投与後一定期間、慎重な避妊措置を講じる必要があります。
脊髄性筋萎縮症に対する理学療法
理学療法は、脊髄性筋萎縮症患者の複合治療とリハビリテーションの基盤の一つとして用いられます。この治療の主なポイントは以下のとおりです。
- サスペンションシステムによる荷重軽減、能動‐受動トレーニング、脊髄の経皮電気刺激の使用。
- 呼吸法と理学療法
- 30 分の垂直化セッション。
- 経言語的電気刺激治療(細かい運動能力を向上させる運動と組み合わせた 20 分間のセッション)
- 手動テクニック;
- さまざまな関節グループへのパラフィンの塗布。
- 筋肉のパフォーマンスを向上させるダーソンバル。
ダーソンバル法は、高電圧・低強度の高周波交流パルス電流を用いて組織に作用させる方法です。一連の施術後、筋力の向上、微小循環の強化、細動脈と毛細血管の拡張、虚血の除去、筋肉への栄養と酸素供給の改善が見られ、再生および萎縮過程に良い影響を与えます。
脊髄筋萎縮症の患者にとって最も重大な問題の一つは呼吸筋の衰弱であり、多くの場合、呼吸機能障害や患者の死につながります。
脊髄性筋萎縮症では、呼吸を司る筋肉を含む骨格筋全体の機能が低下します。筋力低下と緩やかな筋萎縮は呼吸機能の質に悪影響を及ぼし、合併症の発症や呼吸不全の悪化につながります。そのため、筋肉を強化し、呼吸器合併症や呼吸器感染症を予防するための対策を講じる必要があります。その中で特に重要なのが、アンビューバッグを用いた体操です。アンビューバッグは、理学療法、ストレッチ、マッサージと併用して行います。アンビューバッグを使用することで、胸郭と肺の容積を「拡張」することができます。小児の活動には、少なくとも1.5リットルの容量があり、過剰な圧力を解放するバルブ(気圧外傷の予防)を備えたバッグが適しています。
満腹状態では運動を行わないでください。体位は、座位、半座位、横臥位、仰臥位(痰に問題がない場合)。毎回異なる体位で処置を行うのが最適です。患者の背筋をまっすぐにすることが重要です。必要に応じてコルセットを使用します。処置を開始する前に、気道に痰がないことを確認してください。
脊髄性筋萎縮症に対するマッサージ
脊髄性筋萎縮症の治療におけるマッサージは、軽やかで優しいものでなければなりません。筋肉の抵抗がある部位にはタッピングなどの一般的なマッサージを施し、神経支配が保たれている部位には深いストローク(縦方向、横方向)や揉みほぐしを行います。
一般的に、病気の経過や年齢といった個々の特性に応じて、様々な種類のマッサージを実施します。マッサージには以下のようなものがあります。
- 深部の筋肉を刺激するための揉みほぐし。
- 血液とリンパの循環を最適化するためのマッサージ。
- トリガーポイントのスポット治療。
- 繊維強化叩きの。
問題領域全体に効果が広がることが重要です。
脊髄性筋萎縮症に対するマッサージの禁忌:
- 急性炎症、体温上昇;
- 血液疾患、出血傾向;
- 化膿性プロセス;
- 感染性、真菌性皮膚疾患;
- 血管瘤、血栓炎、動脈内膜炎、リンパ節炎;
- 良性および悪性の腫瘍。
脊髄筋萎縮症の患者に対するマッサージは、患者ごとに厳密に処方されます。不適切な施術、過度に乱暴な施術、不適切な刺激は、患者の状態に悪影響を及ぼす可能性があります。
防止
現在、直接・間接DNA診断、そして出生前DNA診断が積極的に進められています。これにより、病気の赤ちゃんが生まれる可能性が大幅に低減されます。これは、脊髄性筋萎縮症のお子さんを出産した経験のあるカップルにとって特に重要です。
予防措置は重要な医療動向を表しており、一次措置、二次措置、三次措置に分類されます。
一次対策は、不利な要因の影響を直接的に防ぎ、病気の進行を防ぐことを目的としています。こうした予防には、食生活や生活習慣の改善、健康的なライフスタイルの実践が含まれます。
二次予防は明らかな危険因子の排除から成り、病状の早期診断、動態の監視の確立、標的治療が含まれます。
三次予防は、特定の運動能力が損なわれた病人に対して行われます。この場合、薬物療法、心理療法、社会療法、労働リハビリテーションが対象となります。
世界保健機関(WHO)の情報によると、世界では2%以上の赤ちゃんが何らかの発達障害を持って生まれています。同時に、これらの障害の0.5~1%は遺伝性です。こうした問題の予防は、医学的な遺伝カウンセリングと質の高い出生前診断に集約され、遺伝病理学的な疾患を持つ赤ちゃんを出産するリスクを最小限に抑えることができます。
脊髄性筋萎縮症やその他の遺伝性疾患を発症するリスクは、両親から受け継いだ遺伝子に依存します。遺伝的要因を早期に特定し、遺伝的に決定された病態の個人リスクを計算することは、標的を絞った予防策と言えるでしょう。
出生前診断には、直接的な調査方法と間接的な調査方法があります。まず、間接的な出生前診断が必要な女性を特定します。これには以下のようなものがあります。
- 35歳以上の妊婦
- 過去に2回以上の自然流産を経験したことがある人。
- 遺伝的発達障害を持つ子供を持つ人。
- 不利な遺伝歴を持つ;
- ウイルス感染または放射線被曝の経験がある方(妊娠計画段階を含む)。
予防目的では、超音波検査やホルモン検査(生化学スクリーニング)などの方法が用いられます。絨毛膜生検、羊水穿刺、胎盤穿刺、臍帯穿刺といった侵襲的な処置も行われる場合があります。遺伝的リスクに関する信頼できる情報を得ることで、生活習慣や妊娠を調整し、病気の子どもの出産を防ぐことができます。
脊髄性筋萎縮症ワクチン
もちろん、脊髄性筋萎縮症のお子様を持つ親御さんは皆、この病気を完全に治したいと願っています。しかし、この病気を根絶できるワクチンは存在しません。最適な治療法の研究は継続して行われています。
特に、2016年にアメリカの科学者が独自の薬剤であるスピンラザ(ヌシネルセン)を承認し、その後、ヨーロッパ諸国でも使用が承認されました。
専門家は脊髄性筋萎縮症の治療問題を以下の方法で調査しています。
- 「間違った」SMN1 遺伝子を修正または置換する。
- 正常なSMN2遺伝子の機能の増強;
- SMNタンパク質欠乏により影響を受ける運動神経細胞の保護。
- 病状の進行を背景に、筋肉の萎縮変化から保護し、失われた機能を予防または回復します。
遺伝子治療では、損傷した遺伝子を標的とし、血液脳膜を通過して脊髄の適切な部位に到達するウイルスベクターを使用します。ウイルスは、遺伝子の欠陥を「縫合」するかのように、損傷した細胞に正常なDNA部分を「感染」させます。こうして運動神経細胞の機能が修復されます。
もう一つの方向性は、SMN2遺伝子の機能を高めることを本質とする低分子療法です。脊髄性筋萎縮症と診断された乳児は、SMN2遺伝子を少なくとも1つ持っています。この方向性はアメリカの科学者によって積極的に研究されており、現在、SMN2遺伝子から完全なタンパク質の合成を促進することを目的とした複数の薬剤が臨床試験中です。
治療介入のもう一つの方法として、運動ニューロンの死を減らし、適応能力を高め、機能性を改善するための神経保護を検討することが挙げられます。
3つ目の方向性は、筋萎縮の進行から筋を保護することです。SMNタンパク質の欠乏は運動神経細胞と筋機能に悪影響を及ぼすため、この治療の目標は、筋萎縮を防ぎ、筋量を増加させ、筋機能を回復させることです。このタイプの治療は遺伝子装置には影響を与えませんが、脊髄性筋萎縮症の悪化を遅らせ、あるいは阻止する可能性があります。
脊髄性筋萎縮症のスクリーニング
新生児スクリーニングは医療現場でますます利用され、しばしば決定的な役割を果たしています。脊髄性筋萎縮症を可能な限り早期に発見することで、患児の予後を大幅に改善することができます。スクリーニング診断には、表に概説されている以下の点が含まれます。
脊髄性筋萎縮症の一種 |
症状学 |
脊髄性筋萎縮症I型(座ることができない、平均余命は最長2年) |
出生後から生後6ヶ月まで症状が現れます。筋緊張の不足、泣き声の弱さ、筋力低下(咀嚼筋や嚥下筋を含む)の増加が見られます。頭位保持に問題があり、横になる際に「カエル」のような姿勢をとります。 |
脊髄性筋萎縮症 II 型(子供は座ることができ、平均余命は通常 2 年以上で、患者の半数以上が 20 ~ 25 歳まで生きます) |
生後7ヶ月から1歳半の間に発症します。嚥下障害、呼吸障害、咳嗽などの症状が現れることもあります。永続的な症状としては、筋肉のけいれん、関節可動域の制限、脊柱の湾曲、低血圧、筋力低下などが挙げられます。 |
脊髄性筋萎縮症III型(座ったり動いたりすることはできるが、上記の能力は徐々に失われ、寿命は正常) |
1歳半で発症します。脊柱と胸郭の湾曲、骨盤と下肢近位部の筋萎縮、関節可動域の増大が認められます。嚥下困難が見られます。 |
脊髄性筋萎縮症IV型 |
成人型を指します。症状は脊髄性筋萎縮症III型と多くの共通点があります。筋力低下は徐々に進行し、振戦や筋線維束性収縮が16~25歳頃に現れます。 |
予測
ウェルドニッヒ・ホフマン症候群の平均余命は1.5~2年です。ほとんどの場合、呼吸不全の進行と肺の炎症の進行により致命的な結果となります。人工呼吸器による適切な呼吸補助により、新生児の余命をわずかに延ばすことができます。継続的な緩和ケアが特に必要であり、これは脊髄性筋萎縮症II型にも不可欠です。III型およびIV型の病態は、より良好な予後を特徴としています。
脊髄性筋萎縮症は、どのタイプでも深刻な病気です。患者の家族全員が、継続的な心理的、情報的、そして社会的サポートを必要とします。患者は、小児科医、神経科医、呼吸器科医、心臓専門医、整形外科医、理学療法士などの専門医による適切な診断と専門的なサポートを受けることが重要です。この疾患に対する特異的な治療法はありませんが、対症療法、特別な栄養(非経口および経腸)の処方、そして病状の進行を遅らせ、合併症の発症を防ぐための様々なリハビリテーション対策が行われます。
多くの患者は障害認定を受けており、個別のリハビリテーション計画が策定されています。
呼吸や摂食を補助するための特別な装置を使用しないで自然に発症した脊髄性筋萎縮症では、約半数の症例で、病気の子どもが 2 歳になる前に死亡します (ほとんどが I 型疾患)。