
骨関節炎の病因の遺伝的および代謝的側面
最後に見直したもの: 23.04.2024

すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
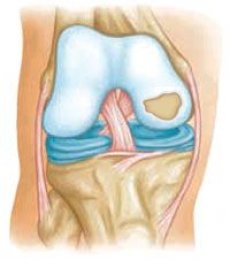
骨関節炎の病因における機械的要因の役割は疑いがありませんが、骨関節炎のいくつかの形態がメンデルの法律に従って継承されているという説得力のあるデータがあります。遺伝性骨関節症は、
- 原発性全般骨関節症(PGOA)、
- 結晶関連関節症、
- 遺伝性骨軟骨異形成症による早期変形性関節症。
Heberdenは1803年に、ブラシの遠位指節間関節の背面にある「やや高密度の結び目、小さなエンドウの大きさ」を説明しました。この特徴は、著者によれば、変形性関節症を痛風を含む他の関節疾患と区別する。J. Hayagarth(1805)は、Geberdenの節の臨床的記述を拡大し、他の言語化の関節症との頻繁な関連性に注目した。Bouchardは、手の近位指節間関節の背側表面上の同様の節をさらに説明した。W. Osierは、 "Heberden and Bushard nodes"という用語を使用して、 "肥大性関節炎"と "変形性関節炎"(1909)を共有しました。1953年、RM StecherとH. Hershは、家族の間でHeberdenの節の分布を発見し、それらが常染色体優性の様式で遺伝すると結論づけた。RM StecherとH. Hershの発見に続いて、GeberdenとBushard節と他の関節の変性病変との関連が明らかになった。JS Lawrence(1977)、JS Lawrenceおよび共同研究者(1983)は、現在の臨床検査およびHLAタイピングに基づいて、単一遺伝子の欠損ではなく多遺伝子遺伝の存在を示唆している。
遺伝性変形性関節症の表現型スペクトルは、成人期に達した後にのみ臨床的に現れる軽度の形態から、小児期に現れる非常に重篤な形態に広範に変化する。伝統的に、これらの形態は全て二次性変形性関節症に分類されている。現在では、これらの表現型のいくつかのための遺伝子発現の軟骨基質の完全性と軟骨細胞の増殖および規制を破壊関節軟骨ECMの巨大分子をコードする遺伝子の変異であることが知られています。これらの遺伝性疾患は、二次的変形性関節症とは異なる骨関節炎の特定の亜群を表す。
遺伝性および続発性の変形性関節症の差異(Williams CJおよびJimenez SA、1999による)
|
遺伝性変形性関節症 |
二次性変形性関節症 |
|
|
病因 |
関節軟骨で発現される遺伝子の突然変異 |
様々な遺伝病および後天性疾患 |
|
病因 |
関節軟骨の構造的または機能的構成要素への損傷 |
関節軟骨のみに常に影響を及ぼすわけではないが、この疾患の二次症状 |
|
治療 |
遺伝子欠損の是正のための遺伝子療法の可能性 |
基礎疾患の治療 |
軟骨異形成/骨軟骨異形成症 - 関節軟骨および成長プレートの成長および発生異常を特徴とする臨床的に異種の疾患群。一部のHD / OXDは臨床的に重度の経過を特徴とする骨関節炎の早期発症につながる。その中で、以下の疾患を区別することができます:
- 脊椎脊髄性異形成(SED)、
- シンドロームStickler、
- 異形成、
- 複数の心理学的異形成(MED)、
- 形而上的軟骨異形成(MHD)、
- oto-spondylo-metaepiphysial dysplasias(OSMED)のいくつか。
変形性関節症の早期発症を特徴とする遺伝性異形成(Williams CJ and Jimenez SA、1999)
|
病気 |
軌跡 |
継承型 |
変異した遺伝子 |
突然変異型 |
|
SED(SAR)の遅発性の早期OA * |
12q13.1-q13.2 |
FROM |
COL 2 A、 |
塩基の置換、挿入、欠失 |
|
Stickler症候群(STL1) |
12q13.1-q13.2 |
FROM |
COL2A1 |
ベースの交換、挿入 |
|
Stickler症候群(STL2) |
6r21.3 |
FROM |
コラ |
挿入、削除 |
|
スティラー症候群 |
1月21日 |
FROM |
コラ |
ベースを交換する |
|
ワーグナー症候群 |
12q13.1-q13.2 |
FROM |
COUA、 |
ベースを交換する |
|
OSMED |
6r21.3 |
AR |
コラ |
ベースを交換する |
|
マーシャル症候群 |
1月21日 |
FROM |
コラ |
挿入 |
|
異形成症の下痢 |
12q13.1-q13.2 |
FROM |
コラ |
挿入、削除 |
|
M3fl(EDM1) |
1913.1 |
FROM |
COMP |
ベースを交換する |
|
BIP(EDM 2) |
1p32.2-rZZ |
FROM |
コラ |
挿入 |
|
MHDShmida(MCDS) |
6q21-q22.3 |
FROM |
コラ |
塩基の置換、欠失 |
|
MXDヤンセナ(MCDJ) |
X21.221.3 |
FROM |
PTHR、 |
ベースを交換する |
*カッコ内は軌跡の記号です。AD - 常染色体優性; ARは常染色体劣性である。
脊椎脊椎動脈異形成
Spondiloepifizialnye異形成(DMS)は、多くの場合、小人症を引き起こし、軸骨格の異常な発達及び長骨の骨端の重い変化によって特徴付けられる常染色体優性遺伝を伴う疾患の不均一な群を含みます。しばしば、SEDは臨床的に進行しにくく、身体の短縮と四肢の縮小を伴う。
後期に現れるSEDの形態では、表現型はほとんど変化しないことが多く、重度の変形性関節症が発症する青年期までは臨床的には見られないことがある。腰椎の変形は、椎間板の狭小化、扁平上皮症および重要でない脊柱後弯症によって明らかにされ得る。また、末梢関節における骨端の異常およびそれらの早期退化性変化がある。末梢関節の病変の最も恒常的な徴候は、大腿骨の顆頭間溝の平坦化と同様に、足関節および膝関節の関節表面の平坦化である。思春期に現れる股関節の骨関節炎の発症に伴って、大腿骨の頭部および頸部の異常がしばしばある。
硝子軟骨のECMの主成分、EDSの原因が遺伝子COL1Aをコードしていることが示唆されている-により、コラーゲンII型は事実です。後半マニフェストSEDに関連した初期の変形性関節症の表現型、およびプロコラーゲンタイプII遺伝子COL間の遺伝的リンクの最初の記述2 Aは、最初の変異COLが報告された1989年と1990年に適用される2 A、後半のマニフェストに関連付けられている初期の変形性関節症と親戚でSED塩基Arg519> Cysの置換に関する。これまでに、類似の突然変異を有する4つの家族が同定されている。このファミリーのメンバーでSED-表現型はEDS連れのご家族の代表はまた、他の変異を発見した位置519のシステインへのアルギニンの交換と家族の表現型に類似していないものの、早期OAと簡単に流れるEDSと他の家族のメンバーは、塩基置換Arg75>のCysを検出しましたCOL 2 A-Gly976 Ser、Gly493 Ser。J.シュプランガーら(1994)は、軟骨一次変異II型プロコラーゲン遺伝子COL1Aを遺伝性疾患を説明するために用語「タイプ11 kollagenopatii」を用います。
Sticklerの症候群の古典的な形
それは1965年にGBスティラーと共著者によって初めて報告されました。彼らは遺伝性関節症 - 眼眼症と呼ばれました。この症候群で記述されたGB症候群は、視力臓器への損傷と、通常は生後3〜4年で発症する重篤な変性性関節疾患によって特徴付けられた。これは常染色体優性疾患であり、その罹患率は新生児1万人あたり約1人である。この病気の臨床像には、近視、進行性難聴、口蓋裂、下顎低形成(Pierre-Robin's anomaly)および脱毛症低形成が含まれる。新生児期には、Sticklerの症候群の患者のX線写真上に、近位大腿骨および遠位脛骨を中心とする拡大骨端が存在する。成長過程において、骨端の異形成が発生し、これは骨端の骨化の不規則性およびその後の変性変化に現れる。
COLため2 A、外観に関連するこの遺伝子スティックラー症候群の病態に、関節軟骨、及び眼球の硝子体中で発現されます。ただし、スティックラー症候群のいくつかのファミリーの調査は、疾患がCOLに関連付けられていないすべての家族ことが示されている2タイプIスティックラー症候群(シンボルSTL1座)と呼ばれる疾患のこの形態A.。
Sticklerの症候群の臨床症状のスペクトルは大きく異なり、現在、いくつかの表現型が同定されている。その中には、眼球の敗血症の有病率を特徴とするワーグナー症候群; ワーグナー症候群のOAは、事実上の変異を持つ患者が特定されて遺伝子COLものの、開発決して2 A、(塩基置換Gly67> ASP)。そのような突然変異COLが硝子体の機能のみを損ない、硝子軟骨に影響を与えない理由は不明である。
Sticklerの症候群の別の形態は、いわゆるオランダの変種である。それは、視覚器官への損傷を除いて、すべての古典的な症候群の症状によって特徴付けられる。HGブルナーら(1994)は、変異COL遺伝子に関連するオランダのスティックラー症候群の表現型を示し、2:優性突然変異ら(1998)エクソンM.Sirko-Osadsaの削除続いて54塩基対の欠失である別のファミリーが報告同様の表現型および遺伝子突然変異COLと、以前の著者とは無関係な説明、及び2(27塩基対の欠失)、データHGブルナーら(1994)をサポートしています。このオプションは、タイプIIスティックラー症候群(シンボルSTL1軌跡)と呼ばれます。
最近、Stickler症候群の3番目の座位が、硝子体病変および網膜病変を有する家族で見られ、表現型的には、この症候群の「古典的」バージョンで観察された変化とは著しく異なる。このファミリーの代表者は、遺伝子COL2Aの突然変異を発見した| (塩基Gly97> Valの置換)。もちろん、AJ Richardsと共同研究者の所見を確認するために、Sticklerの症候群のこのような表現型および遺伝子型の症例の新しい記述が必要である。
長い間、マーシャル症候群とSticklerの症候群の古典的なバージョンのnosological接続の問題が議論されました。今や、マーシャル症候群は、主に顔面骨格のより顕著な変形のために、別個の表現型に分類されるが、末梢関節損傷は、I型症候群と同様である。マーシャル症候群では、30年後に膝関節および腰仙脊椎の変形性関節症が始まります。この症候群の原因は、タイプCOL n A1のコラーゲンIX遺伝子の突然変異である。
OSMED
この表現型は、変形性関節症は思春期に表示され、主に影響を与え、腰、膝、肘や肩の関節に似た、オランダの家族の中で関節内のメンバーの退行性変化を説明しました。また、しかし、ビジョンのいずれかの異常の臓器を明らかにしなかった、独特の顔の特徴を見つけ、難聴、腰椎前弯、増加した節間関節を増加させた(Vikkula M.ら、1995)。研究者たちは、コードする遺伝子の変異を発見した2 -鎖タイプIIコラーゲンCOLを,, 2。
異形成症の下痢
それは、胴体と四肢の短縮、鼻の顔面と背部の平坦化、眼球の眼球の外転および関節の重度の異常を特徴とする。Knin症候群の患者では、通常は出生時の関節が小児期および早期の青年期に増加し続ける。彼らはまた、しばしば近視、難聴、口蓋裂、内反足を検出することができます。患者の大部分は、特に膝および股関節で発現される早期の重篤な変性変化を発症する。脊柱のレントゲン写真上では、椎体の平坦化およびかなりの伸長、扁平上皮症が検出される。長い管状の骨はダンベルとして変形し、骨端の骨化は遅くなる。手の関節では、骨端が平坦化され関節ジョイントが狭められる。関節軟骨は柔らかく、弾力性は低下する。組織学的には、大きなシスト(「スイスチーズ」の症状)がその中に見出される。Knyst症候群の原因は、COb2A1型のプロコラーゲンII遺伝子の変異である。
 [7], [8], [9], [10], [11], [12], [13], [14]
[7], [8], [9], [10], [11], [12], [13], [14]
複数の骨端性異形成(MED)
軸方向および末梢関節(通常膝、腰、肩、手の関節)の両方に影響を与える長骨の成長板の異常な発達、ならびに早期(マニフェスト小児期における)重度の変形性関節症によって特徴付けられる疾患の不均質なグループ。臨床的には、MEDは関節における痛みおよび堅さ、歩行の変化によって明らかになる。DERの患者はまた時々バックボーン無傷で、(椎体の平坦化の様々な程度の)脊柱における最小限の変化を示します。小人症はめったに発生しないが、患者の低成長も特徴的である。ビジョンの器官は影響を受けません。DERにはいくつかの変異体、例えばFerbanksおよびRibbingの表現型が含まれる。
MEDは浸透度の異なる常染色体優性型で遺伝する。DER異常骨端成長板の顕著な特徴であるので、これらの遺伝子の原因が不良異形成エンコード高分子軟骨成長板であることが示唆されています。少なくとも3つの遺伝子座がDER表現型と関連していることが判明した。Research E.J. ウィーバーら(1993)、「犯人」DER遺伝子から除外JTヘクトおよび共同研究者(1992)はタイプIIおよびVI、プロテオグリカンおよび軟骨のタンパク質カップリングのコアタンパク質を型コラーゲン。JTヘクトおよび共同研究者(1993)、R. Oehelmannら(1994)は、シンドロームpsevdoahondroplaziiおよび染色体19の動原体周囲領域これDERと臨床的に近いとの間の関連を見出しました。その後の研究は、EDRを有する3人の患者(座位記号EDM1)における軟骨オリゴマーマトリックスタンパク質(OMPC)をコードする遺伝子の突然変異を同定した。すべての3つの変異はカルシウム結合ドメインOMPHをコードする遺伝子の領域で発生しているので、おそらくこのタンパク質のカルシウム結合機能は、軟骨成長板の正常な発達のために必須です。
MDブリッグスら(1994)は、IX型コラーゲンCOL1A1(EDM座シンボルの遺伝子の一つを含む、第1染色体の部分と関連していたオランダの家族、DER-表現型を報告した2)。観測された変異は硝子軟骨の整合性を維持するには、コラーゲン線維IIの表面にコラーゲンタイプIXの役割の最初の証拠ローカライズされたことは注目に値します。ディアM.ら(1995)フェアバンクスの表現型は、遺伝的に任意の軌跡EDM ,,ない軌跡EDMに関連付けられていないことを示した2 DERの不均一性を確認しました。
形而上性軟骨異形成(MHD)
異種臨床早期変形性関節症が現れる硝子軟骨の遺伝性障害の群、(150の以上の種類を説明します)。MCHは骨幹端骨の変化を特徴とする。臨床的に、彼らは低身長、短い足、下肢の曲率、「鴨」歩行を明示する。また、MHDを有する患者は、他の系(例えば、免疫系および消化系)に損傷の徴候を示す。軟骨成長板の観察された崩壊は、組織学的に明らかなクラスタが増殖及び肥大軟骨細胞、増粘隔壁及び無秩序マトリックス、および軟骨下骨に浸透nekaltsifitsirovannogo軟骨に囲ま。
Jansen、SchmidおよびMcCusickの症候群は、最もよく研究されたMHDである。それらは骨格異常の特徴において同様であるが、重症度が異なる(Jansen症候群 - McKusick症候群-Schmid症候群)。最も一般的なものは、常染色体優性型に遺伝するSchmid症候群(MCDS座の象徴)である。X線症候群は、管状骨の短縮および湾曲、象形質のカップ様変形(遠位大腿骨よりも近位でより顕著である)、凝固斑によって現れる。最も顕著な変化は、長い管状骨の成長プレートで観察される。
Schmid症候群の患者には、X型コラーゲン遺伝子の少なくとも17の異なる変異が記載されている。コラーゲンX型は、増殖プレートの肥大化軟骨細胞において発現され、おそらくは骨化過程に関与する。したがって、COb2A1遺伝子のコラーゲンをコードするX型の変異は、シュミット症候群の最も原因である可能性が高い。
ヤンセン症候群の小児では、高カルシウム血症、尿中のリン酸塩レベルの上昇、副甲状腺ホルモン(PG)およびPG結合ペプチドのレベルの低下があります。後者の異常、おそらく、ジャンセンの症候群の出現。1994年、AS Karaplisと共同研究者が元の研究結果を発表した。マウス胚の幹細胞中のPG結合ペプチドをコードする遺伝子が破壊された後、この対立遺伝子を欠損したマウスは出生直後に死亡する。彼らは、軟骨下骨の発達、軟骨の成長の侵害、および軟骨細胞の増殖の低下に異常を有していた。1995年、E. Schipaniおよび共同研究者らは、Jansen症候群の患者におけるPG結合型ペプチド受容体遺伝子のヘテロ接合突然変異を報告した。突然変異は、cAMPの蓄積をもたらすGys223> Argの塩基を置換することからなり、これは、223位のアミノ酸ヒスチジンがシグナル伝達において重要な役割を果たすことを意味する。後のE. Schipaniおよび共同研究者(1996)は、Jansen症候群の患者3人を報告し、そのうち2人は同様の変異を有し、3人目はTruA10> Proを置換した。
原発性全般性変形性関節症
遺伝プライマリ一般変形性関節症の最も頻繁な形態は、最初の臨床プライマリ一般骨関節炎特性外観ブシャールノードとheberden、多関節病変別nosology JH Kellgren R. Mooreのように1952年に記載された変形性関節症(アフリカマイニングパートナーシップ)、です。プライマリ一般変形性関節症、変形性関節症は、彼の進行の急速な症状の早期発症によって特徴付けられます。X線写真で主要一般変形性関節症は、非遺伝性の変形性関節症と異なりません。主な一般化変形性関節症の病因の質問はまだ議論されているという事実にもかかわらず、研究が主な一般的な変形性関節症の発生および進行における遺伝的素因の重要な役割を実証しています。
数字はそれぞれ17および一般集団の26%であったので、JH Kellgrenら(1963)は、男性親族の36%、及び女性親族の49%にBusharai heberdenノードを発見しました。プライマリ一般変形性関節症を有する患者において、ますますHLAハプロタイプA1V8とMZ-アイソフォームA1アンチトリプシンを見つけます。双子TDスペクターら(1996)の古典的な研究では膝のX線および変形性関節症の特徴的な変化が存在する単一130および二卵性双生児120ツイン女性の手の関節を行います。すべてのサイトが二卵性双生児と遺伝的要因と比べて一卵性双生児で2倍高かった変形性関節症の一致放射線証拠ということを発見した40〜70%の範囲であったからに貢献。(1997)GDライトらを行った研究結節性変形性関節症は、患者の年齢や概念の両親に疾患、重症度および疾患の発症年齢との間に高い負の相関関係の早期発症を実証しました。
結晶関連関節症の中で、関節腔における尿酸およびカルシウム含有結晶の結晶の堆積は、家族の素因を有する。
遺伝性結晶関連性関節症(Williams、C.J. And Jimenez SA、1999による)
|
病気 |
軌跡 |
継承型 |
変異した遺伝子 |
突然変異型 |
|
痛風(HPRT)* |
Xq27 |
X染色体に関連する |
HPRT1 |
塩基の置換、欠失 |
|
痛風(PRPS) |
Xq22-q24 |
X染色体に関連する |
PRPS1 |
ベースを交換する |
|
一次ピロリン酸関節症(CCAL1) |
5r15.1-r15.2 |
FROM |
? |
? |
|
0Aの早期発症に関連するピロリン酸性関節症(CCAL2) |
8q |
FROM |
? |
? |
*カッコ内は軌跡の記号です。ADは常染色体優性である。
D. Zintann S. Sitajは1958年に27人の患者に「軟骨石灰化症」と呼ばれる病理学の臨床記述を発表しました。ほとんどの患者は5家族に属し、病気の病因発生に遺伝的要素を示した。その後、D. McCartyとJL Hollander(1961)は、関節腔に非永久結晶が沈着しているとの痛風を疑う2人の患者を報告した。X線検査では、多くの関節の硝子軟骨の異常な石灰化が明らかになった。
X線撮影カルシウムピロリン酸関節症の疾患ピロリン酸二水和物結晶沈着または散発OAに似ているが、しかし、それはしばしば関節に影響を与え、従来のフォームosteoartrozaa(例えば、中手指節、舟状ビーム、膝蓋骨 - 大腿膝部)のための典型的ではありません。ピロリン酸関節症は、多くの場合、軟骨下骨嚢胞を形成した場合。ほとんどの場合、二次変形性関節症の徴候の前に起こる軟骨が、いくつかの個体において、疾患は代謝の障害を伴う特発性変形性関節症、として開始することができる(血色素症、副甲状腺機能亢進症、gipomagnezemiyaら)。
ほとんどの場合、関節軟骨のECMの構造変化は、ピロリン酸カルシウム水和物の結晶の析出を誘発します。AO ピロリン酸関節症のコラーゲン含有量の減少とコラーゲン線維の断片化とスウェーデンの関節軟骨マトリックスファミリーの中央ゾーンで見つかったBjelle(1972、1981)。これらのサイトは、結晶を含んでいなかったので、著者らは述べた行列異常が彼らの堆積や関節の退行性変化の発展に素因得ることを示唆しました。ピロリン酸関節症K.イシカワら(1989)、I.増田ら(1991)の散発性症例の研究に基づいて原因の軟骨は、ECMのタンパク質をコードする遺伝子の突然変異であると結論付けました。CJWilliamsら(1993)、AJ Reginatoら(1994)は、ヘテロ接合変異COLた2(拠点置き換え、AをArgl5>のCys) ankilozirovaniya伴う重度の初期の変形性関節症の臨床表現型を持つ大家族のメンバー、遅発性spondiloepifizialnoy異形成と硝子の軟骨と繊維を軟骨。しかし、それはこの家族の軟骨のメンバーはOAとの関係で二次的自然を身に着けていたことが判明しました。
結晶の形成は、ECMの無機成分によって促進されることも示唆された。例えば、低マグネシウム血症は、結晶の溶解を減少させるピロホスファターゼ酵素を阻害することによって軟骨石灰化の発症を引き起こす。ピロリン酸関節症の患者の滑液において、無機リン酸塩の含有量の増加が検出された。この観察および他の観察により、ピロリン酸関節症の患者には、ピロリン酸の代謝に局所的な障害が存在することが示唆された。おそらくECM中のそれらの沈着ゾーンにおいてピロリン酸塩結晶の形成に関与するヌクレオシド三リン酸ピロホスホヒドロラーゼの酵素が記載されている。散発性のケースでは、ピロリン酸関節症は、酵素の増加内容を観察したが、そのような疾患、異常の家族型は観察されなかった(ライアンLMら、1986)。しかし、家族性ピロリン酸関節症の患者の培養線維芽細胞及びリンパ芽球はまた、疾患の病因における局所的な代謝障害ピロリン酸の役割についての仮説を支持する無機リン酸、レベルの上昇を検出しました。
近年では、試みは、家族の場合にピロリン酸関節症の原因で、「有罪」の遺伝子を決定するために行われました。だから、厳しい急速に進行する変形性関節症nedisplasticheskomuへの二次開発軟骨れるピロリン酸関節症と大家族のメンバー(メイン州、USA)から入手した遺伝物質の分析は、病気の軌跡COLとの接続を除外2。しかし、この研究の著者らは、8番染色体の長腕(軌跡シンボルSSAL)上に位置する、研究された表現型ピロリン酸関節症と軌跡との間の関連を見出しました。AGヒューズら(1995)は、領域5r15に第5染色体の短腕上に局在している英国と軌跡CCAL1 ,,から家庭内のプライマリ軟骨の表現型の間の関連を見出しました。前のケースよりもピロリン酸関節症、いくつかのローカライズされた近位端とアルゼンチンから家族にCJ Williamsら(1996)、CCAL1軌跡によって出願された- 5r15.1領域に。同様の遺伝子型がフランスの家族で見つかった。
したがって、記載された研究のデータは、ピロリン酸関節症のファミリー形態が、少なくとも3つの異なる遺伝子の突然変異によって引き起こされる臨床的および遺伝的に異種の疾患であることを示している。