
アッシャー症候群
最後に見直したもの: 04.07.2025

すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
アッシャー症候群は、出生時から完全な難聴を呈し、加齢とともに進行する失明を呈する遺伝性疾患です。視力低下は網膜色素変性症(網膜の色素変性症)と関連しており、多くのアッシャー症候群患者は重度の平衡感覚障害も呈します。
疫学
研究の結果、検査対象となった聾唖児の約8%がアッシャー症候群に罹患していることが判明しました(検査は聾唖者のための専門施設で実施されました)。先天性難聴の患者の6~10%に色素性網膜炎が認められ、色素性網膜疾患の患者の約30%に認められます。
この疾患は、世界中で10万人あたり約3~10人に発症すると考えられています。男女ともに発症率は同程度で、世界人口の約5~6%がこの症候群に罹患しています。小児重度難聴の症例の約10%は、アッシャー症候群I型およびII型が原因です。
米国では、1型と2型が最も多くみられます。これらを合わせると、小児のアッシャー症候群の症例全体の約90~95%を占めます。
原因 アッシャー症候群
アッシャー症候群のI型、II型、III型は常染色体劣性遺伝による原因で、IV型はX染色体異常と考えられています。この症候群に伴う失明や難聴の原因は、まだ十分に研究されていません。この疾患の患者は、DNA構造を損傷する可能性のある成分に対して過敏症を示すと考えられています。また、この疾患は免疫系疾患と関連している可能性がありますが、この場合、その過程の正確な解明は進んでいません。
1989年、II型疾患の患者において初めて染色体異常が特定されました。これは将来、この症候群の原因遺伝子を単離する方法につながる可能性があります。また、保因者におけるこれらの遺伝子を特定し、特別な出生前遺伝子検査を開発することも可能になるかもしれません。
 [ 8 ]
[ 8 ]
症状 アッシャー症候群
アッシャー症候群の症状には、難聴や眼構造における色素細胞の異常な蓄積などがあります。その後、網膜変性が進行し、視力低下を引き起こし、最悪の場合、失明に至ります。
感音難聴は軽度または完全難聴の場合があり、通常は出生時から進行することはありません。しかし、網膜色素変性症は小児期以降に発症する可能性があります。検査結果によると、周辺視力が低下しても(「トンネル視野」と呼ばれる状態)、中心視力は長年維持できることが示されています。
これらが病気の主な症状であり、精神病やその他の精神障害、内耳の問題、白内障などの他の障害が加わることもあります。
フォーム
研究中に、この病気の 3 つのタイプと、非常にまれな 4 番目のタイプが特定されました。
I型は、先天性完全聾と平衡障害を特徴とします。多くの場合、このタイプの子供は1歳半になってようやく歩き始めます。視力の低下は通常10歳から始まり、最終的に夜盲症へと進行するのは20歳頃です。このタイプの病気を持つ子供は、周辺視力が徐々に低下していくことがあります。
II型では、中等度または先天性の難聴が認められます。この場合、部分的な難聴の悪化はしばしば起こらなくなります。色素性網膜炎は、思春期の終わり頃、または20歳を過ぎた頃から発症し始めます。夜盲症の発症は通常29~31歳で始まります。II型病変の視力障害は、一般的にI型よりもややゆっくりと進行します。
タイプ III の病気は、通常は思春期に始まる進行性の難聴と、同じ時期 (難聴より少し遅れて) に網膜色素変性症が徐々に進行することを特徴としており、これが進行性の失明の要因となることがあります。
IV型病変の症状は主に男性に現れます。この場合、進行性の障害や聴覚および視覚の喪失も観察されます。この病型は非常にまれで、通常はX染色体性です。
診断 アッシャー症候群
アッシャー症候群の診断は、患者に観察された突発性難聴と進行性視力喪失の組み合わせに基づいて行われます。
テスト
変異を検出するために特別な遺伝子検査が指示されることがあります。
アッシャー症候群の発症を引き起こす可能性のある遺伝子座が 11 個発見されており、この障害の明確な原因となる遺伝子が 9 個特定されています。
- タイプ 1: MY07A、USH1C、Cdh23、Pcdh15、SANS。
- タイプ 2: ush2a、VLGR1、WHRN。
- アッシャー症候群3型:USH3A。
NIDCDの科学者たちは、ニューヨークとイスラエルの大学の同僚と共同で、ユダヤ人集団における1型アッシャー症候群の大部分を占めるPcdh15遺伝子のR245Xと呼ばれる変異を特定した。
臨床試験を実施する研究所について知るには、https://www.genetests.org にアクセスし、研究所のディレクトリで「アッシャー症候群」を検索してください。
アッシャー症候群の遺伝子検査を含む既存の臨床試験について詳しくは、https://www.clinicaltrials.gov にアクセスし、「アッシャー症候群」または「アッシャー症候群の遺伝子検査」を検索してください。
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
機器診断
機器診断にはいくつかの方法があります。
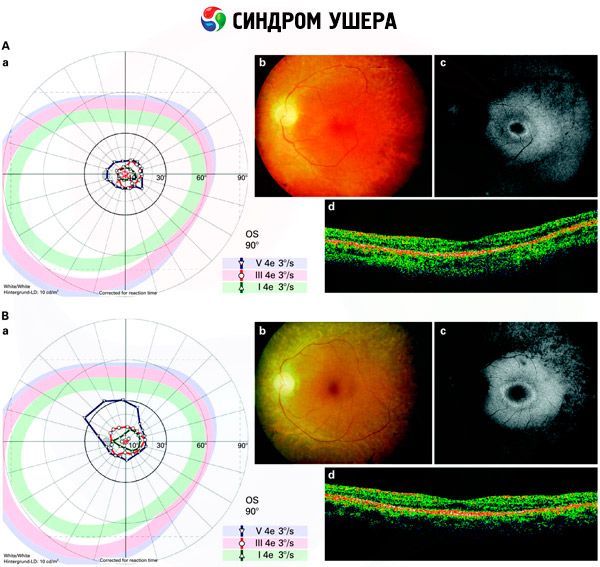
- 網膜上の色素斑の存在や網膜血管の狭窄を検出するための眼底検査。
- 網膜電図は、網膜における初期の変性異常を検出することができます。この検査では、放射線電図上の伝導路の消失が示されます。
- 電気眼振図(ENG)は、不均衡の存在を示す可能性のある不随意眼球運動を測定します。
- 聴力検査は難聴の有無とその重症度を判断するために使用されます。
差動診断
アッシャー症候群は、いくつかの類似した疾患と区別する必要があります。
ハルグレン症候群は、先天性の難聴と進行性の視力低下(白内障と眼振も併発)を特徴とする疾患です。その他の症状としては、運動失調、精神運動障害、精神病、知的障害などが挙げられます。
アルストロム症候群は、網膜が変性し、中心視力の喪失につながる遺伝性疾患です。この症候群は小児期の肥満と関連しており、同時に10歳を過ぎると糖尿病と難聴が発症し始めます。
妊娠初期の女性が風疹に罹患すると、胎児の発育に様々な異常が生じる可能性があります。こうした異常の結果には、難聴や視覚障害(または視覚障害)、そして様々な発達障害などがあります。
連絡先
処理 アッシャー症候群
現在、アッシャー症候群を完治させる治療法はありません。そのため、この疾患の治療は主に視力低下の進行を遅らせることと、聴力低下を補うことに重点が置かれます。考えられる治療法には以下のものがあります。
- ビタミン A の摂取(一部の眼科医は、ビタミン A パルミチン酸エステルの大量摂取は網膜色素変性症の進行を遅らせることはできるが、止めることはできないと考えています)。
- 患者の耳に特殊な電子機器を埋め込むこと(補聴器、人工内耳)。
眼科医は、一般的な網膜色素変性症の成人患者の大部分に対し、監督下でビタミンAパルミチン酸エステルを1日15,000IU(国際単位)摂取することを推奨しています。本研究ではアッシャー症候群1型患者は含まれていなかったため、この患者群には高用量のビタミンAは推奨されません。ビタミンAの摂取を検討している方は、この治療法について医師にご相談ください。この治療法に関するその他の推奨事項は以下のとおりです。
- ビタミン A を多く含む食品を食生活に取り入れる。
- 妊娠を計画している女性は、先天異常のリスクが高まるため、妊娠予定日の3か月前に高用量のビタミン A の摂取を中止する必要があります。
- 妊娠中の女性は、先天異常のリスクが高まるため、高用量のビタミン A の摂取を中止する必要があります。
このような子どもを社会生活に適応させることも重要です。そのためには、特別支援教育の教師や心理学者の支援が必要です。患者が視力低下の兆候を示し始めた場合は、手話の使用を指導する必要があります。
予測
アッシャー症候群の予後は不良です。この疾患のどのタイプにおいても、ほとんどの患者において、視野と視力は20~30歳の間に悪化し始めます。場合によっては、両眼の視力が完全に失われることもあります。難聴は必ず吃音を伴い、急速に両眼の完全な難聴へと進行します。