
クラッベ病
最後に見直したもの: 29.06.2025

すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
クラッベ病は、ガラクトシルセレブロシド(GALC)欠損ガラクトシダーゼとしても知られ、リソソーム病のグループに属する希少遺伝性疾患です。この遺伝性疾患は、GALC遺伝子の変異によって引き起こされ、その結果、体内のガラクトシルセレブロシダーゼ酵素が欠乏します。
クラッベ病は中枢神経系と神経を侵し、組織内に有害物質が蓄積します。症状には、発達遅延、技能の喪失、筋力低下、発作、視覚障害および聴覚障害、進行性障害などがあります。クラッベ病は重篤な疾患であり、多くの場合、生活の質の低下、そして最終的には死に至ります。
この病気は遺伝性で、幼少期または成人期に発症する可能性があります。診断は通常、臨床症状、遺伝子検査、生化学検査に基づいて行われます。
現在、特異的な治療法はありませんが、早期発見と対症療法の継続は患者さんの生活の質の向上につながります。新たな治療法の開発に向けた研究と臨床試験が現在も行われています。
疫学
クラッベ病の疫学は世界各地で多様です。この疾患は稀であり、その有病率は民族や地域によって大きく異なります。
- 世界的に:クラッベ病はまれな遺伝性疾患と考えられており、世界中での正確な有病率は不明です。
- 地理的差異:有病率は国や地域によって異なる場合があります。一部の民族集団では、有病率が高くなる場合があります。
- 遺伝:クラッベ病は常染色体劣性遺伝であり、両親から子に欠陥遺伝子が伝わらないとその病気を発症しません。両親が変異遺伝子を保有している必要があるため、この病気は稀です。
- スクリーニングと診断:一部の国では、症状が現れる前であっても新生児のクラッベ病を検出できる新生児スクリーニングプログラムを実施しています。これは、地域によって疾患の検出率に影響を及ぼす可能性があります。
原因 クラッベ病
クラッベ病は異染性白質ジストロフィー I 型としても知られ、14 番染色体の GALC (ガラクトース レブロシダーゼ) 遺伝子の変異によって引き起こされる遺伝性疾患です。この疾患は、酵素ガラクトース レブロシダーゼの欠乏または完全な活性の欠如によって引き起こされます。
通常、ガラクトセレブロシダーゼは、神経細胞のミエリン鞘に蓄積するガラクトセレブロシドの分解に関与しています。このガラクトセレブロシドの蓄積は、ミエリン(神経線維の絶縁体)の破壊につながり、運動機能や協調運動障害、発達遅延、神経系の機能低下といったクラッベ病の症状を引き起こします。
クラッベ病は常染色体劣性遺伝により家族内で遺伝するため、両親がGALC遺伝子の変異を有する場合にのみ、子供がこの病気を発症するリスクが高くなります。両親が両方ともこの変異を有する場合、その子は変異遺伝子を2つ受け継ぐ確率が25%です。
病因
クラッベ病は、ガラクトセレブロシダーゼ(GALC)という酵素の欠損によって引き起こされる遺伝性神経変性疾患です。この酵素は、神経系における脂質(脂肪)の代謝、特に神経線維を絶縁するミエリンを構成するスルファチドの分解に重要な役割を果たしています。
クラッベ病の病因は、ガラクトセレブロシダーゼ欠損によるスルファチド分解阻害にあります。この阻害により、神経細胞、特にミエリン合成に重要な役割を果たすオリゴデンドロサイトにスルファチドが蓄積します。オリゴデンドロサイトやその他の神経細胞へのスルファチドの蓄積は、炎症とミエリンの変性を引き起こし、神経線維の損傷と神経線維死を引き起こします。
ミエリンと神経細胞の徐々に進行する破壊は神経系機能の低下につながり、運動能力の変化、筋力低下、協調運動障害、技能喪失、認知機能および精神機能障害といった症状として現れます。症状の重症度と病状の経過は、個々の症例およびGALC遺伝子の変異によって異なります。
症状 クラッベ病
クラッベ病の症状には、患者の生涯にわたって発現する様々な兆候が含まれます。症状は、病気の形態や重症度によって異なります。以下にその一部をご紹介します。
乳児および幼児(病気の初期段階)の場合:
- スキルの喪失と発達の退行。
- 低緊張(筋力低下)。
- 摂食および嚥下の問題。
- 這う、座る、歩く能力の喪失を含む運動能力の変化。
- 視覚と聴覚機能の変化。
- 発作。
年長児および青年(発症が遅い場合)の場合:
- 進行性の運動機能低下。
- 運動協調の悪化。
- 言語発達の遅れ。
- 知的障害を含む認知障害。
- 日常の活動におけるスキルと自立性の喪失。
- 発作。
- 精神的および感情的な問題。
成人(クラッベ病の成人型)の場合:
- 進行性の運動障害。
- 歩行能力および自己管理能力の喪失。
- 視覚と聴覚の問題。
- 認知障害および精神障害。
- 周囲の世界とコミュニケーションをとり、理解する能力が徐々に失われます。
症状は時間の経過とともに次第に重くなり、通常は障害を負い、最終的には死に至ります。
ステージ
クラッベ病の一般的な段階は次のとおりです。
初期段階:この段階は通常、乳児期に始まります。症状には以下が含まれます。
- スキルの喪失と発達の退行。
- 低緊張(筋力低下)。
- 摂食および嚥下の問題。
- 這う、座る、歩く能力の喪失を含む運動能力の変化。
- 視覚と聴覚機能の変化。
- 発作。
中期:病気が進行するにつれて、症状はより顕著になり、より広範囲の機能に影響を及ぼします。この段階は小児期または青年期に始まり、以下の症状が含まれます。
- 進行性の運動機能低下および運動協調の喪失。
- 言語発達の遅れと認知障害。
- 独立した活動や自己管理を行う能力が徐々に失われます。
- 発作。
- 精神的および感情的な問題。
後期:この段階では、症状がさらに重症化し、完全な障害に至ります。思春期または成人期に発症する可能性があり、以下の症状が含まれます。
- 進行性の運動障害と歩行能力の喪失。
- 自己管理能力の喪失および日常活動における自立性の喪失。
- 視覚と聴覚の問題。
- 認知障害および精神障害。
- コミュニケーション能力と周囲の世界を理解する能力の喪失。
フォーム
クラッベ病には次のようないくつかの種類があります。
- 乳児型:最も一般的で重篤な病型です。症状は通常、乳児期(通常3~6ヶ月)に始まります。乳児型の小児では、発達退行、運動能力の低下、筋緊張低下(筋力低下)、摂食障害、発作がみられ、最終的には運動機能と社会性を失うことがあります。病気の進行は通常急速で、ほとんどの小児は成人まで生存できません。
- 若年型:この型は通常、小児期(思春期に入って数年後)に発症します。症状は乳児型よりも軽度で進行も遅い場合もありますが、運動能力の低下、発達の退行、認知障害、その他の神経症状が依然として見られます。
- 青年期型および成人型:これらの型はまれで、青年期または成人期に発症します。症状には、進行性の運動機能低下、うつ病や精神病などの精神状態の変化、認知障害などがあります。病気の進行は緩やかで、中年期、あるいは老年期まで生存する患者もいます。
合併症とその結果
合併症は、症状の発症年齢や病気の種類によって異なります。クラッベ病の典型的な合併症には、以下のものがあります。
- 運動能力の喪失:子どもは筋肉を動かしたり協調させたりする能力を失います。その結果、運動能力と移動能力が制限されます。
- 発達の退行: ほとんどの子供は発達の退行を経験します。これは、以前に獲得したスキルや能力を失うことを意味します。
- 認知障害:クラッベ病は、記憶障害、知能低下、学習障害などの認知障害を引き起こす可能性があります。
- 言語およびコミュニケーション障害: 子どもは言語およびコミュニケーション能力を発達させる能力を失う可能性があります。
- てんかん発作:一部の患者ではてんかん発作が起こる場合があります。
- 呼吸困難および摂食困難: 病気が進行すると呼吸困難および摂食困難に陥り、人工呼吸器や栄養チューブの使用などの医療サポートが必要になる場合があります。
- 精神的および感情的な問題: 患者はうつ病や攻撃性などの精神的および感情的な問題を経験する場合があります。
- 寿命の短縮: より重篤な場合、子供は通常は成人まで生きられず、平均寿命は子供時代に限られます。
診断 クラッベ病
クラッベ病の診断にはいくつかの方法と手順があります。
- 臨床評価:医師は身体診察を行い、家族歴を含む病歴を収集し、病気の兆候と症状を特定します。運動能力の低下、発達の退行、行動やコミュニケーションの変化などの症状が認められる場合、クラッベ病の存在が疑われることがあります。
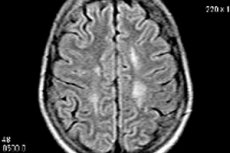
- 神経画像検査:診断を確定するために、磁気共鳴画像法(MRI)やコンピュータ断層撮影法(CT)などの神経画像検査が必要になる場合があります。これらの検査により、疾患の特徴となる可能性のある脳の変化を視覚化することができます。
- 生化学検査:診断のために、血液と尿の生化学検査を行い、疾患に関連する特定の代謝物や酵素の有無を調べます。これには、酵素ガラクトセブロシダーゼの活性分析が含まれる場合があります。
- 遺伝子検査:最後に、診断を確定するために遺伝子検査が行われます。この検査では、疾患の根本原因であるGALC遺伝子の変異の有無が確認されます。遺伝子検査は、症状のある小児、または遺伝子変異を受け継ぐリスクのある家族のいずれかに対して実施できます。
診断は多くの場合、特徴的な症状や検査結果の変化が認められる幼少期に行われます。早期発見により、患者さんの生活の質の向上を目的とした治療と支持療法を開始することができます。
差動診断
クラッベ病の鑑別診断には、クラッベ病に類似する可能性のある徴候や症状を特定するとともに、他の疾患と鑑別するための検査や診察を行う必要があります。以下に、その一部をご紹介します。
- その他のリソソーム病:クラッベ病は、ムコ多糖症やゴーシェ病などを含むリソソーム病群の一つです。鑑別診断では、生化学検査や遺伝子解析に基づいて他のリソソーム病を除外する必要があります。
- 脊髄性筋萎縮症(SMA):これは遺伝性の神経変性疾患群であり、筋力低下や運動能力の低下といった類似の症状を呈することがあります。鑑別診断には、SMAの特定の病型を特定するための遺伝子検査が含まれます。
- 脳性麻痺(脳性麻痺):この疾患は、筋力低下や運動機能障害を伴う点でクラッベ病と類似することがあります。鑑別診断は、臨床症状と神経画像検査に基づいて行われます。
- 神経軸索ジストロフィー:これはまれな神経変性疾患であり、クラッベ病に類似した症状を呈することもあります。鑑別診断には生化学検査と遺伝子検査が含まれます。
正確な診断を下し、同様の症状を示す可能性のある他の病気を除外するためには、遺伝学者や神経学者を含む医療専門家による包括的な検査と相談が必要です。
処理 クラッベ病
クラッベ病の治療は依然として困難であり、この病気を完全に治癒する特異的な治療法は存在しません。しかし、症状を緩和し、生活の質を向上させるための治療と維持には様々なアプローチがあります。以下にそのいくつかをご紹介します。
- 骨髄移植:早期診断と適切な介入が行われた場合にこの治療法が用いられます。骨髄移植は病気の進行を遅らせる可能性がありますが、必ずしも効果的ではなく、深刻なリスクや合併症を伴う可能性があります。
- 対症療法:主な焦点は病気の症状の緩和です。これには理学療法、言語療法、発作、痛み、その他の症状を抑える治療が含まれます。
- 支持療法: 患者には、生活の質を向上させるための看護、身体のリハビリテーション、専門的なサービスなど、さまざまなサポートが提供される場合があります。
- 研究: 遺伝子治療法や生物学的医薬品など、クラッベ病の新しい治療法の開発に向けた研究が進行中です。
治療は患者様一人ひとりのニーズに合わせて個別化され、個々のニーズに基づいて行われるべきです。クラッベ病は患者さんの生活に大きな影響を与える可能性があるため、ご家族や介護者へのサポートも重要です。患者さんとそのご家族には、医師やリハビリテーション専門家と協力して、生活の質を最大限に高め、症状を緩和することが推奨されます。
防止
クラッベ病はまれな遺伝性疾患であるため、予防措置は、子供がこの病気を持って生まれるのを防ぐことを目的としており、遺伝のリスクがある家族向けに設計されています。
- 遺伝カウンセリング:クラッベ病の遺伝リスクが高いカップルは、妊娠を計画する前、または妊娠初期に遺伝カウンセリングを受けることができます。遺伝カウンセラーは、クラッベ病の遺伝の可能性や、妊娠を希望する親のためのスクリーニングや診断方法について情報を提供することができます。
- 遺伝子変異のスクリーニング:家族歴や他の遺伝性疾患がある場合、妊婦に対して遺伝子変異のスクリーニングが行われます。これは、子孫への疾患の遺伝リスクを特定するのに役立ちます。
- 周産期検査:クラッベ病の遺伝リスクが高い夫婦は、子供への病気の遺伝を防ぐための選択肢を検討する場合があります。一つの選択肢として、ドナー精子や卵子を用いた人工授精などの生殖補助医療の利用が挙げられます。
- 胎児遺伝子検査:妊婦がクラッベ病の遺伝リスクが高い場合、胎児の出生前遺伝子検査が行われることがあります。これにより、妊娠継続の判断を早期に下せる可能性があります。
- ライフスタイルと健康: クラッベ病のリスクを軽減するための特別な予防策はありませんが、妊娠中は健康的なライフスタイルを維持し、赤ちゃんの健康に影響を与える可能性のある要因を避けることが常に推奨されます。
予測
クラッベ病の予後は、病気の種類、重症度、そして症状が初めて現れた年齢によって異なります。特に早期に診断・治療された場合、ほとんどの患者さんの全体的な予後は以下のようなものとなります。
- 早期診断と治療:早期診断と治療(通常は生後数ヶ月以内)は、予後を大幅に改善することができます。骨髄移植などの治療法は、病気の進行を遅らせ、平均余命を延ばす可能性があります。
- 重症型:重症型、特に幼少期またはそれ以降に症状が発現した患者は、予後が不良となる可能性があります。これらの病型は、重度の神経系障害と寿命の短縮につながる可能性があります。
- 支持療法: クラッベ病の管理において重要な側面は支持療法であり、これには身体のリハビリテーション、専門的なトレーニング プログラム、社会的支援などが含まれます。
- 個々の予後:予後は常に個別であり、患者の特性、疾患の重症度、治療の有効性によって異なります。軽症で早期に診断された患者は、重症で遅く診断された患者よりも予後が良好となる可能性があります。
Использованная литература