記事の医療専門家

新しい出版物
クモ足
最後に見直したもの: 04.07.2025

すべてのiLiveコンテンツは、可能な限り事実上の正確さを保証するために医学的にレビューまたは事実確認されています。
厳格な調達ガイドラインがあり、評判の良いメディアサイト、学術研究機関、そして可能であれば医学的に査読された研究のみにリンクしています。 かっこ内の数字([1]、[2]など)は、これらの研究へのクリック可能なリンクです。
当社のコンテンツのいずれかが不正確、期限切れ、またはその他の疑問があると思われる場合は、それを選択してCtrl + Enterキーを押してください。
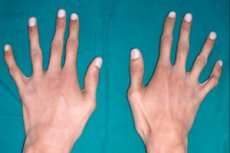
まれな遺伝性の結合組織病変の 1 つにクモ指症があります。これは指の変形で、管状骨の伸長、骨格の湾曲、心血管系および視覚器官の障害を伴います。
この疾患は、ウィリアムズ博士とフランスの小児科医マルファンによって初めて報告され、彼らはこれを長指症(dolichostenomelia)と名付けました。その後、マルファン症候群という用語が世界の医学界に登場し、1902年以降はクモ指症(arachnodactyly)という名称が使われるようになりました。この病理は結合組織の先天性疾患に関連しており、主な兆候の一つは、長く、薄く、湾曲した「クモ指」と考えられています。[ 1 ]
疫学
クモ指症は、常染色体優性遺伝、高い浸透度、およびさまざまな程度の表現度を伴う、結合組織の遺伝性単一遺伝子病変であると考えられています。
約75~80%の症例では遺伝性であり、残りの症例では自然発生的な突然変異(特に点ミスセンス変異)の結果として発生します。[ 2 ]
クモ指症の病因的特徴は、いくつかの遺伝子の変化によって引き起こされることが証明されています(症例の 95%)。
- 染色体15q21.1上のフィブリリン-1遺伝子(約1000個の変異が研究されている)
- 9番染色体および3p24.2-P25にあるTGFβR1またはTGFβR2遺伝子。
これらの変化は臨床症状の程度に直接影響を及ぼします。
I 型コラーゲンの α-2 鎖の変異が患者の 5% で報告されています。
クモ指症の有病率は5000人あたり約2人です。人種や性別による分類はされていません。[ 3 ]
原因 クモダクティ
クモ指症は自然突然変異の結果として発症します。この疾患は、先天性遺伝病に一般的に見られる徴候を示さず、指の伸長は主にマルファン症候群とホモシスチン尿症で発現します。
この疾患は稀であり、発見された患者全員に15番染色体のq21.1領域に位置するFBN1遺伝子(フィブリリン遺伝子)の変異が認められた。この症候群の臨床的多型性は、変異の多さによって説明でき、そのうち約15%は受胎中に生じた変異である。[ 4 ]
クモ指症の非定型的な種類は、他の遺伝子の変異によって引き起こされます。病状の発症と、染色体 14 の q24 領域に位置し成長因子を変換する LTBP3、R 遺伝子の変異との間に関係が発見されています。
変異の圧倒的多数は、いわゆるミスセンス変異(約57%)です。20%以上は染色体部分の喪失を伴う小さな欠失、約12%はスプライシング部位の変異、8%はナンセンス変異、2%は大規模な再編成と欠失です。[ 5 ]
先天性拘縮性クモ指症は、タンパク質因子フィブリリン-2の合成障害によって引き起こされます。この疾患は、第5染色体のq23-q31領域に位置するFBN2遺伝子の変異によって引き起こされます。これらの変異は主に点突然変異であり、コドンが別のアミノ酸をコードするように変化します。[ 6 ]
ホモシスチン尿症は、必須アミノ酸であるメチオニンの代謝障害を背景に発症します。病態の根本原因としては、以下の酵素物質の活性低下または減少が考えられます。
- シスタチオニンβシンターゼ(CBS)酵素。この病態はビタミンB6抵抗性であり、ビタミンB6非依存性型の症状を呈しますが、ビタミンB6製剤の導入によって改善は得られません。
- 酵素N5、N10メチレンテトラヒドロ葉酸還元酵素(MTHFR)。この酵素剤は、ホモシステインの脱メチル化と、多くのタンパク質やペプチド物質の成分となる必須アミノ酸であるメチオニンへの変換を引き起こす基質です。
- 酵素N5メチレンテトラヒドロ葉酸。これはビタミンB6の欠乏によって発症する、ビタミンB6依存型の病態です。ビタミン剤を体内に摂取することで、患者の症状を改善することができます。
- ホモシステイントランスメチラーゼ酵素。コバラミン(ビタミンB12 )の代謝障害を背景に発症します。
危険因子
クモ指症は、遺伝学的に決定的な疾患であり、結合組織の全身的損傷を特徴とします。この病態は、15番染色体短腕の21.1座に位置するフィブリリン1遺伝子の変異に関連しています。
クモ指症は常染色体優性遺伝で、浸透率が高く、表現度は様々です。この病態は男性側と女性側の両方に発現する可能性があります。
病因
人体の質量の 50% 以上は、骨格、皮膚、血管網、リンパ、血液などの結合組織構造で構成されています。
結合組織細胞は、線維芽細胞とそのサブタイプ(骨芽細胞、軟骨細胞、角質芽細胞、象牙芽細胞)、およびマクロファージと肥満細胞によって代表されます。
胎児期の結合組織は、体質、遺伝的要素、そしてエピジェネティック要素の形成の材料です。結合組織疾患は、程度の差はあれ、全身、その機能的能力、そして体質に反映されます。
クモ指症では、フィブリリン-1ペプチドの構造情報を含む遺伝子のヌクレオチド置換がみられます。このタンパク質物質は糖タンパク質グループに属し、ミクロフィブリル系に関与し、結合組織の弾性線維の基礎を形成します。
組織は細胞間マトリックスによって安定した構造を維持しており、細胞間マトリックスには多くの成長因子が含まれており、系統的な細胞再生を促します。大血管や靭帯には多数のエラスチン線維が含まれており、その損傷がクモ指症の主な症状を引き起こします。
形質転換成長因子βは著しく損傷を受け、その不活性型は結合されず、この因子の生物学的活性が増加します。
線維性疾患は線維の異常な形成を引き起こし、皮膚やその他の結合組織構造の強度と弾力性の低下を引き起こします。
コラーゲン構造が破壊されると、止血における主要なつながりが破壊されます。
専門家は、クモ指症を含む結合組織構造の異常の発生と進行にホルモンの不均衡が関与していることを指摘しています。血栓症の症状は、特に腕頭動脈領域の血管の血液粘度の変化といったレオロジー異常によって決定されます。
消化管はコラーゲン障害により影響を受けます:低運動性胆道ジスキネジア、横隔膜の食道開口部のヘルニア、肝胆道異常、慢性型の「消失した」胃十二指腸炎、長嚢胞状奇形が観察されます。
クモ指症はどのように遺伝するのでしょうか?
クモ指症は常染色体優性遺伝形式をとりますが、自然突然変異によっても発生することがあります。つまり、この病態は必ずしも近親者(例えば両親や祖父母)に発現するとは限りません。病態の根底にある突然変異は、偶然に明らかになることもあります。
症状 クモダクティ
医学では、潜在性クモ指症と顕性クモ指症を区別しています。潜在性クモ指症では、変化は見られますが、それらは孤立しており、1つか2つの器官系に限定されています。顕性クモ指症は、症状の強さは様々ですが、複数の目に見える障害として現れます。顕性クモ指症の場合、患者の状態は数十年間比較的安定している場合もあれば、他の器官や器官系との関連で徐々に進行する場合もあります。
多くの場合、クモ指症は赤ちゃんの誕生後数日以内に検出されます。
クモ指症の主な外的兆候は、細く長い指であり、腱が短いため、典型的な湾曲を呈し、クモの脚に似ています。
クモ指症、つまりクモ指症は、新生児期からはっきりと現れます。しかし、3歳頃になると、症状はより顕著になります。
成長指標にも特徴が見られます。子供は通常、背が高く、前腕と脛が長く、四肢は不均衡で細いです。この不均衡は、特に腰と膝頭が局所的に高くなっていることで顕著です。[ 7 ]
その他の骨格障害には以下のものがあります:
- 異常な顔面領域を伴う、狭く長い頭蓋骨の形状(長頭症)
- 胸部が漏斗状になっている(いわゆる「鳥」)。
- 湾曲した背骨;
- 先天性股関節脱臼;
- 習慣的な膝肩関節脱臼;
- かかと骨の湾曲;
- 骨棘形成 – 骨の成長;
- 扁平足;
- 脂肪層の発達が不十分。
クモ指症のその他の兆候としては、次のようなものがあります。
- 近視;
- 青みがかった強膜;
- 水晶体の亜脱臼;
- 心臓や血管の障害(心臓欠陥、大動脈瘤)
- 顕著な薄さ。
- 関節過剰可動性;
- 「アーチ状の」空。
クモ指症は、細長い管状骨を特徴とし、様々な骨格の湾曲を引き起こします。さらに、多くの患者は、正常な精神発達を背景に、錐体路障害、腱反射の非対称性、瞳孔閉鎖、眼振などの症状を示します。
ホモシスチン尿症を伴うクモ指症の場合、患者は次のような症状を経験する可能性があります。
- 周期的なけいれん発作;
- 水晶体亜脱臼を伴う続発性緑内障;
- 網膜剥離、乱視;
- 動脈幹(腎臓、脳を含む)の損傷
- 骨粗鬆症;
- 血栓症;
- 知的障害。
新生児のクモ指症
ほとんどの場合、クモ指症は生後数日以内に小児に発症します。しかし、年齢を重ねるにつれて症状は悪化する傾向があります。
通常、この病気が疑われる最初の兆候は次のとおりです。
- 異常に長い手足と指。
- 子供の適切な身体的状態における低体重;
- 細長い顔。
- 体脂肪の不足、筋肉の発達不良;
- 関節の過度の柔軟性。
4歳頃から、クモ指症を背景に、胸部が変化し始め、脊柱が湾曲し、扁平足が発達します。
視覚器官に関しては、近視、水晶体の偏位、角膜の形状変化、斜視、虹彩および網膜の形成不全などが観察されます。これらの障害は、生後2~3歳で既に認められますが、年齢を重ねるにつれて進行します。
最も危険なのは心血管系の障害です。このような障害がある場合、適切な治療を受けなければ、小児期に既に死亡するリスクが高くなります。特に危険な病態としては、血管壁の損傷、心臓奇形、冠動脈の欠損などが挙げられます。場合によっては、生後1年目の乳児において、心不全の進行が既に認められることがあります。
クモ指症は、他の臓器、特に神経系、気管支、肺、皮膚、泌尿生殖器系の損傷を伴うことがあります。これらの障害は診断検査中に発見されます。
合併症とその結果
クモ指症の最も一般的な副作用は、心臓と呼吸器官への損傷です。さらに、視力障害、場合によっては視力喪失も起こります。
クモ指症の患者は、既存の病態すべてに対する複合的な治療を必要とします。このような治療が行われない場合、患者の平均余命は著しく短縮します。医療介入を受けずに40歳まで生きる患者は稀です。年月が経つにつれて、心血管系の問題は悪化し、悲惨な結果をもたらす広範囲の出血のリスクが高まります。
クモ指症に罹患している人は、定期的に検査を受け、付随する病状を治療する必要があります。この状態でのみ、深刻な合併症を起こさずに長生きできる可能性が高くなります。
マルファン症候群およびクモ指症の女性患者が妊娠した場合、特別な注意が必要です。このような状況では、患者を定期的かつ徹底的に検査し、心血管系の状態を医学的にサポートすることが重要です。これが、妊娠の成功と安全な出産を保証する唯一の方法です。
クモ指症の患者は、副作用を避けるため、定期的に医師の診察を受け、心血管疾患や筋骨格系の疾患の発症を防ぐ必要があります。また、適度な運動を含む、健康的なライフスタイルを送ることが推奨されます。
診断 クモダクティ
クモ指症の臨床診断には、症状の聴取、家族歴、表現型評価、人体計測および身体検査が含まれます。直系血縁者における結合組織病変の有無を確認することが重要であり、そのために系図学的研究が行われます。[ 8 ]
クモ指症の臨床診断には、骨や軟骨組織、血液、リンパ液といった特定の結合組織の状態を調べることが含まれる。最も有益なのは、1日尿中のオキシプロリンとグリコサミノグリカンの濃度を調べることであると考えられている。さらに、血液中のリジン、プロリン、オキシプロリンの含有量も評価する。異なるタイプのコラーゲンの比率の変化やコラーゲン繊維の構造異常を判断するために、コラーゲンとフィブロネクチンに対するポリクローナル抗体を用いた間接蛍光抗体法による分類が処方される。[ 9 ] 適応症に応じて、結合組織の代謝プロセスの質を反映するために、以下の検査が実行される。
- グリコサミノグリカン、フィブリリン、フィブロネクチン;
- ヒドロキシプロリン、1 型コラーゲン生合成マーカー、1 型プロコラーゲンのアミノ末端プロペプチド、1 型コラーゲン分解マーカー、ガラクトシルオキシリジン、デオキシピリジノリンの研究。
- コラーゲン代謝調節の分析(マトリックスメタロプロテアーゼおよびマトリックスメタロプロテアーゼの組織阻害剤、形質転換成長因子の研究)
- 微量元素および多量元素の分析(カルシウム、マグネシウム、リン、鉄、硫黄、銅、コバルト、セレン、亜鉛、マンガン、フッ素、バナジウム、ケイ素、ホウ素の含有量)
- 骨組織形成のマーカーとリモデリング過程の速度の研究(尿と血液中のオステオカルシン、骨アルカリホスファターゼ、副甲状腺ホルモン、成長ホルモン、プロラクチン、ビタミンD3 、ペントシジン、ホモシステインの含有量の分析)。
機器診断は、既存の発達障害を特定し、臓器および器官系の機能状態を評価することを目的としています。クモ指症における心血管疾患は、以下の検査を診断リストに必須に含める必要があります。
- 心電図検査;
- 24時間心電図モニタリング;
- 心臓と血管の超音波検査。
血管異常は、CT(コンピュータ断層撮影)またはMRI(磁気共鳴画像)を用いて検査されます。追加の診断検査には、以下のようなものがあります。
- 腹部臓器、泌尿生殖器系の超音波検査。
- 胸部臓器、股関節のX線写真;[ 10 ]
- 脊椎のCTまたはMRI。
必要な検査がすべて終了したら、患者は遺伝カウンセリングを受けるよう紹介されます。
差動診断
ビールズ症候群とマルファン症候群のクモ指症(それぞれFBN2遺伝子とFBN1遺伝子の変異)の間には表現型の類似性があり、これはタンパク質物質フィブリリン1とフィブリリン2がほぼ完全に同一であるためです。ビールズ症候群の別名が拘縮性クモ指症であることは驚くべきことではありません。[ 11 ]
他の結合組織病変との鑑別も行われます。
- スティックラー症候群;
- エーラスダンロス症候群;
- ホモシスチン尿症;
- 関節拘縮。
ビールズ症候群は、マルファン症候群様表現型、大小関節の先天性屈曲拘縮、手関節および足部のクモ指症、脊椎の湾曲、そして耳介の形状異常(いわゆる「しわ耳」)を特徴とします。診断確定のため、FBN2遺伝子の分子解析が行われます。
関節拘縮症は、多関節拘縮、低身長、知能低下を特徴とする。指や耳の形状は特筆すべきものではない。[ 12 ]
エーラス・ダンロス症候群は、脊柱後弯症、胸部の漏斗状の弯曲、顕著な扁平足、屈折異常を特徴とする。可動性亢進、関節脱臼、皮膚の伸展性と過敏性が典型的にみられる。
スティックラー症候群は、関節の容積と硬直の変化を特徴とします。
連絡先
処理 クモダクティ
クモ指症は遺伝性疾患であり、現在の医学では遺伝子欠陥を修復する方法が確立されていません。そのため、治療は患者の状態を最適化し、病状の悪化を防ぎ、症状を緩和することを目指します。最も顕著な症状の種類に応じて、複数の専門医による複合的な治療が処方されます。多くの場合、整形外科医、心臓専門医、眼科医、消化器専門医、その他の専門医への相談も必要となります。[ 13 ]
患者の臨床管理に関する推奨事項の中で、一般原則と考えられるのは以下のとおりです。
- 身体活動を制限しますが、完全に排除するわけではありません(心血管系のサポートの一環として)。
- 薬物療法;
- 必要であれば、心臓と血管の最も影響を受けた領域の外科的治療。
- 整形外科的矯正;
- スパトリートメント、理学療法、治療運動。
クモ指症患者の食事には、微量元素、ビタミン、脂肪酸が豊富な高タンパク質食品を十分に含める必要があります。肉、魚、魚介類、豆類、ナッツ類の摂取が推奨されます。クモ指症がホモシスチン尿症によって引き起こされる場合は、動物性タンパク質の摂取を大幅に制限する必要があります。
体格が細く身長が高い子供には、綿実油、大豆油、ヒマワリの種、豚脂、ラードなどを幼い頃から食事に取り入れることが推奨されています。さらに、成長ホルモンの産生を抑制するオメガなどの多価不飽和脂肪酸を含む製剤も与えましょう。
タンパク質代謝を正常化するために、ビタミンB群が処方されます。これらは、そば、エンドウ豆、レバーなどの食品からも摂取できます。
アスコルビン酸を食物を通して体内に取り込むことは非常に重要です。そのためには、ローズヒップの煎じ液、ピーマン、キャベツ、柑橘類、そしてシーバックソーンやネギを食事に必ず含める必要があります。
必要に応じて、脊椎や関節への負担を軽減するために整形外科的矯正が行われます。この目的で、整形外科用靴、膝用装具などの器具、インソール、弾性包帯などが使用されます。
適応に応じて外科的治療が行われます。
薬
クモ指症の薬物治療は、患者の状態と病理学的症状の重症度に応じて、年に1~2回行われます。治療期間は個別に決定され、平均4か月です。[ 14 ]
コラーゲンの生成を刺激するために、以下の薬剤が処方されます:ピアスクレジン 300、L-リジンまたは L-プロリンと、アスコルビン酸、ビタミン B、トコフェロール、マグネシウム、亜鉛、セレン、銅を含む複合マルチビタミン製剤の組み合わせ。
軟骨保護剤の中で最も最適な使用法は、コンドロイチン硫酸とグルコサミン硫酸です。これらは、軟骨細胞の代謝の調節、酵素合成の抑制、酵素作用に対する軟骨細胞の感受性の増加、同化プロセスの刺激などに関与する薬剤です。Teraflex、Arthroflex、Arthraなどの薬剤の併用が最適と考えられています。
ミネラル代謝は、リン-カルシウム代謝を正常化する薬剤によって促進されます。選択される薬剤は、活性型ビタミンD(アルファD3、テバ、オキシデビット、ボンビバなど)であることが多いです。同時に、リン、カルシウム、マグネシウム製剤も使用されます。治療中は、約20日に1回、血中または尿中のカルシウムとリンの濃度を検査し、アルカリホスファターゼの血液検査を行います。
体の生体エネルギー状態を改善するために、ホスファデン、リボキシン、レシチン、エルカー、コエンザイム Q10 を処方することが可能です。
おおよその治療計画は次のようになります。
- 年齢に応じた用量のコンドロプロテクターを、食事と十分な量の水と一緒に服用してください。1回の治療期間は3~4ヶ月です。
- L-プロリン500mg(12歳以上の子供および成人)を、1日1~2回、食前30分に6週間服用してください。必要に応じて、L-プロリン、L-リジン、L-ロイシンを含むアミノ酸複合体を体重1kgあたり10~12mgの割合で追加処方することもできます。服用は1日1~2回、2ヶ月間継続してください。
- ビタミン・ミネラル複合製剤「セントラム」、「ビトラム」、「ユニキャップ」を年齢を考慮した用量で服用してください。投与期間は4週間です。
この治療計画は、クモ指症の患者が筋骨格系の問題を訴え、毎日の尿検査でグリコサミノグリカンの排泄量が増加し、血液中の遊離アミノ酸のレベルが低下していることが検査結果で示されている場合に適しています。
原則として、この治療は患者さんに好意的に受け止められており、特別な副作用もありません。過敏症反応が検出された場合は、薬剤を変更し、治療計画を調整します。
理学療法治療
クモ指症に対する理学療法は、適応症に応じて処方されます。例えば、骨形成が不十分な場合、骨折の治癒を促進する場合、または骨粗鬆症の兆候がある場合は、5%塩化カルシウム、4%硫酸マグネシウム、2%硫酸銅、または2%硫酸亜鉛を用いた電気泳動が推奨されます。
栄養血管障害が検出された場合は、1% カフェイン安息香酸ナトリウム、塩酸エフェドリン、またはメサトンが使用されます。
副腎皮質の機能を活性化させるために、1.5%エチミゾールによる薬物電気泳動と副腎領域へのデシメートル療法が処方される。[ 15 ]
血管の緊張を安定させるには、血管の「体操」を促す水浴が推奨されます。二酸化炭素、松脂、塩化物水素、硫化水素、ラドンなどの入浴法が効果的です。擦り込み、水浴、コントラストシャワー、泡浴、塩浴なども行われます。
軟骨組織の状態を改善するために、磁気療法、誘導療法、レーザー療法、ジメチルスルホキシド(ジメキシド)による電気泳動法が使用されます。
外科的治療
クモ指症に対する外科手術は、適応症に応じて厳密に処方されます。例えば、弁尖脱出を伴う重度の血行動態障害が認められる場合は、巨大大動脈瘤の摘出、弁置換術、および大動脈損傷部の切除が行われます。[ 16 ]
胸部の重度の湾曲の結果として呼吸器系および心血管系の顕著な機能障害が生じた場合は、胸郭形成術が行われます。
3~4度の重度の側弯症によって引き起こされる進行性疼痛症候群の場合も、外科的介入が適応となります。眼科的観点からは、水晶体除去の絶対的な適応は、二次緑内障を伴う水晶体亜脱臼、白内障、および網膜剥離のリスクが高い網膜変性です。
クモ指症やその他の結合組織構造に影響を与える疾患の患者に対する手術は、臨床的および生化学的に比較的症状が緩和した段階でのみ行われます。手術後は、長期にわたる医学的観察と、結合組織代謝を改善する薬剤を用いた集中治療が必須です。
防止
クモ指症はまれな遺伝病です。一見健康な両親に自然発生することもあり、事前に予防することは非常に困難です。
両親のどちらかに重篤な家族歴がある場合、つまり家族にクモ指症の患者がいることが分かっている場合は、子どもを計画する段階で遺伝専門医を訪ね、遺伝子検査を受ける必要があります。妊娠が確認された後、医師は胎児の超音波検査やいくつかの生化学検査を含む出生前診断を行います。
- 母体血液検査;
- 羊水分析;
- 絨毛膜絨毛サンプル採取;
- 胎盤細胞と臍帯血の分析。
クモ指症の患者は生涯にわたって医学的モニタリングを受けます。患者に対する予防措置は、合併症の予防を目的としています。この目的のために、心臓外科手術による矯正が行われ、血栓形成のリスクを排除するための薬物療法が処方されます。抗生物質療法は定期的に実施されます。
予測
クモ指症患者の生命予後は、主に併存疾患、特に心血管系、筋骨格系、視覚器官の疾患の重症度によって決定されます。多くの場合、病理学的には大動脈の破裂や解離が合併します。この点を考慮し、適切な時期に心臓外科手術による治療を行う必要があります。これにより、患者の延命と生活の質の向上につながります。
診断が適切な時期に行われ、医師の勧告に従えば、予後は条件付きで良好と言えるでしょう。医学的サポートにより、クモ指症の経過は大幅に緩和され、患者は深刻な合併症を発症することなく、通常の生活を送ることができ、比較的充実した人生を送ることができます。